分离的双加氧酶及其编码基因与应用
1.本发明涉及一种分离的α-酮戊二酸依赖型双加氧酶tw2odd5及其编码基因,可催化对映-贝壳杉烯酸c-2位羟化,属于药用植物基因工程领域。
背景技术:
2.雷公藤(tripterygium wilfordii hook.f.)是传统中药雷公藤的植物来源,具有清热解毒,祛风除湿,杀虫止痒,消肿散结等功效(高伟,刘梦婷,程琪庆,等.雷公藤的本草考证[j].世界中医药,2012,7(6):560-562.)。现代研究表明雷公藤可用于风湿性关节炎,肾小球肾炎,肾病综合征等病的治疗。萜类成分是雷公藤的主要活性成分,贝壳杉烷型二萜是其中的重要组成部分。从中药的活性成分中开发新药是一种很有前景的方式,然而由于植物的生长缓慢且药用成分含量很低,大大限制了其应用。近年来利用合成生物学技术设计和改造微生物菌株来生产天然活性产物已经成为一种极具潜力的获取方法。
[0003]
萜类化合物在高等植物中的合成途径主要有两种,位于细胞质中的mva途径和位于质体中mep途径,通过以上两个途径生成萜类的共同前体异戊烯基焦磷酸(ipp)及其同分异构体二甲基丙烯基焦磷酸(dmapp);在异戊烯转移酶的催化下,生成ggpp前体,再由二萜合酶(diterpene synthase)催化形成二萜烯中间体母核结构(su p.*,gao l.h.*,liu s.,et al.probing the function of protein farnesyltransferase in tripterygium wilfordii[j].plant cell reports,2019,38(2):211-220),然后母核在氧化酶、甲基转移酶等后修饰酶的作用下生成复杂多样的二萜类活性化合物。
[0004]
α-酮戊二酸依赖型双加氧酶(2-oxoglutarate/fe(ii)-dependent dioxygenases,2-odds),是一类不含血红素的单核铁原子蛋白,与细胞色素p450单加氧酶相比,2-odds反应时仅需要2个或者3个配体,因此具有更高的灵活性,其所催化的反应更具多样性和复杂性(farrow和facchini2014)。2-odds是一个庞大的基因家族,一般根据其氨基酸序列blast p值分为3个具有明显差异的亚家族,分别是doxa、doxb和doxc(kawai等2014),其中doxa和doxb调控植物关键的生长发育过程,如初生代谢、多肽链的翻译后修饰等,在数量和蛋白序列上具有较高的保守性。doxc亚家族参与植物的次生代谢,包括酚酸类,生物碱类以及萜类成分等,它在不同植物体内数量和功能差异较大,具有较大的物种间特异性(kawai等2014)。这三类2-odds在植物界中广泛存在,可以催化羟基化反应、去甲基化反应、开环反应、去饱和反应、闭(成)环反应和卤化反应等(islam等2018)。
[0005]
张逸风等人成功鉴定了雷公藤中的首个α-酮戊二酸依赖型双加氧酶twga13ox,,体外酶促研究证实其可以催化赤霉素a9的c13位发生氧化,生成赤霉素a20。本发明从雷公藤悬浮细胞中克隆得到一条α-酮戊二酸依赖型双加氧酶基因tw2odd5。通过体外酶促反应验证其皆具有催化对映-贝壳杉烯酸(ent-kauranoic acid,缩写ka)的c2位发生氧化生成相应羟化产物的功能。该基因是从雷公藤中首次克隆得到的,在本发明被公布之前,尚未有任何公开或报道过本专利申请中所提及的雷公藤2odd酶基因及其氨基酸序列。
技术实现要素:
[0006]
本发明的第一方面,提供了一种分离的酶,所述酶参与二萜类化合物的生物合成,尤其是贝壳杉烷型二萜类化合物,如2-羟基对映贝壳杉烯酸。
[0007]
本发明中,所述分离的酶,是一种双加氧化镁,尤其是一种α-酮戊二酸依赖型双加氧酶(以下可用tw2odd5表示),为一种参与贝壳杉烷型二萜类化合物的生物合成,尤其是2-羟基对映贝壳杉烯酸合成的关键酶,所述酶具有如seq id no:2所示的氨基酸序列,或seq id no:2所示氨基酸序列经取代、缺失或增加一个或多个氨基酸其功能相同的肽。
[0008]
可以在tw2odd5酶中进行氨基酸序列改变以便最少破坏对生物学活性必须的高级结构。例如,当tw2odd5酶包含一个或多个螺旋时,将改变氨基酸残基以便不破坏螺旋的几何学和其中的构象改变将使一些关键功能(例如分子与其结合伙伴的结合)减弱的其它分子成分。氨基酸序列改变的效果可以通过例如以上公开的计算机模拟进行预测,或通过晶体结构分析进行测定(见例如lapthorn等,nat.struct.biol.2:266-268,1995)。本领域熟知的其它技术对改变蛋白和标准分子(例如天然蛋白)的折叠进行比较,例如,可以比较变体和标准分子中的半胱氨酸分布情况。质谱和使用还原和烷基化的化学修饰为测定与二硫键相关的或未形成这些键的半胱氨酸残基提供了方法(bean等,anal.biochem.201:216-266,1992;gray,protein sci.2:1732-1748,1993;和patterson等,anal.chem.66:3727-3732,1994)。一般认为如果修饰的分子与标准分子具有不相同的半胱氨酸分布情况,则折叠受到影响。另一个用于测量这点的被接受的熟知方法是园二色谱(cd)。测量和比较修饰分子和标准分子产生的cd谱是常规的(johnson,proteins 7:205-214,1990)。晶体学是另一个分析折叠和结构的熟知方法,核磁共振(nmr)、消化肽作图和表位作图也是分析折叠及蛋白质和多肽之间的结构相似性的已知方法(schaanan等,science 257:961-964,1992)。
[0009]
本发明中,tw2odd5酶的变异体形式包括:同源序列、保守性变异体、等位变异体、天然突变体、诱导突变体、在高或低的严谨条件下能与tw2odd5酶编码基因杂交的dna所编码的蛋白。
[0010]
本发明的第二方面,提供了一种编码本发明所述酶的多核苷酸(或称α-酮戊二酸依赖型双加氧酶编码基因,以下可用tw2odd5表示),所述多核苷酸优选具有如seq id no:1所示核苷酸序列及其简并序列。该简并序列指位于seq id no:1序列核苷酸中,有一个或多个密码子被编码相同氨基酸的简并密码子所取代后而产生的序列。由于密码子的简并性,所以与seq id no:1核苷酸序列同源性低至约70%的简并序列也能编码出seq id no:2所述酶的氨基酸序列。还包括能在中度严谨条件下,更佳的在高度严谨条件下与seq id no:1的核苷酸序列杂交的核苷酸序列。还包括与seq id no:1的核苷酸序列的同源性至少70%,较佳地至少80%,更佳地至少90%,最佳地至少95%的核苷酸序列。还包括能编码具有与天然的α-酮戊二酸依赖型双加氧酶相同功能的蛋白的seq id no:1中开放阅读框序列的变异形式。这些变异形式包括(但不限于)若干个(通常为1-90个,较佳地1-60个,更佳地1-20个,最佳地1-10个)核苷酸的缺失、插入和/或取代,以及在5’和/或3’端添加数个(通常为60个以内,较佳地为30个以内,更佳地为10个以内,最佳地为5个以内)核苷酸。
[0011]
本发明中,"tw2odd5"即指α-酮戊二酸依赖型双加氧酶,"tw2odd5"指α-酮戊二酸依赖型双加氧酶编码基因。
[0012]
本发明的第三方面,提供了一种包含本发明第二方面所述多核苷酸的表达载体,
所述的表达载体,如可以是选自peasy系列的表达载体,pet系列表达载体。
[0013]
本发明中,可选用本领域已知的各种载体,如市售的载体,包括质粒、粘粒等。在生产本发明tw2odd5酶时,可以将tw2odd5酶编码基因的核苷酸序列可操作地连接于表达调控序列,从而形成α-酮戊二酸依赖型双加氧酶表达载体。所述“可操作地连连接”当指dna区段时表示这些区段按照一定方式排列以致它们可以协调地为其预期的目的发挥作用,例如在启动子中起始转录并前行通过编码区段到达终止子。也指这样一种状况:即线性dna序列的某些部分能够影响同一线性dna序列其它部分活性,例如,如果信号肽dna作为前提表达并参与多肽的分泌,那么信号肽(分泌前导序列)dna就是可操作地连接接于多肽dna;如果启动子控制序列转录,那么它是可操作地连接于编码序列;如果核糖体结合位点被置于能使其翻译的位置时,那么它是可操作地连于编码序列。一般,“可操作地连接”意味着相邻,而对于分泌前导序列则意味着在阅读框中相邻。
[0014]
本发明第四方面,提供了一种包含本发明第二方面所述多核苷酸、或本发明第三方面所述表达载体的重组宿主菌,所述宿主菌为大肠杆菌,如dh5α菌株、bl21(de3)菌株、bl21(de3)plyss菌株、jm109菌株、hb101菌株等。
[0015]
本发明述宿主菌,包含本发明所述编码tw2odd5酶或其变体的多核苷酸分子,或在严谨条件下可与所述多核苷酸分子进行杂交的核苷酸分子,或包含本发明上述描述的表达载体。所述的宿主细胞选自:细菌、原核细胞(如大肠杆菌、)真菌细胞、酵母细胞、昆虫细胞、哺乳动物细胞或植物细胞,优选地,为大肠杆菌。
[0016]
根据常规方法在含有养分和其它对于所选宿主细胞的生长必须的成分的培养基中培养转化的或转染的宿主细胞。多种适宜的培养基,包括已知成分培养基和复杂培养基,是本领域已知的,一般包括碳源、氮源、必需氨基酸、维生素和矿物质。如果需要,培养基还可以含有诸如生长因子或血清这样的成分。生长培养基一般通过例如药物筛选或缺少可由表达载体携带的或共转染至宿主细胞中的选择标记补充的必需养分来选择含有外源添加dna的细胞。通过常规方式,如振摇小三角瓶或发酵罐喷气给液体培养物提供充足的空气。
[0017]
本发明编码tw2odd5酶的多核苷酸全长序列或其片段通常可以用pcr扩增法、重组法或人工合成的方法获得。对于pcr扩增法,可根据本发明所公开的有关核苷酸序列,尤其是开放阅读框序列来设计引物,并用市售的cdna库或按本领域技术人员已知的常规方法制备cdna库作为模板,扩增而得有关序列。当序列较长时,常常需要进行两次或多次pcr扩增,然后再将各次扩增出的片段按正确次序拼接在一起。一旦获得了有关的序列,就可以用重组法来大批量地获得有关序列。这通常是将其克隆入载体,再转入细胞,然后通过常规方法从增殖后的宿主细胞中分离得到有关序列。此外,还可通过化学合成将突变体引入本发明蛋白序列中。除了用重组法产生之外,本发明蛋白的片段还可用固相技术,通过直接合成肽而加以生产(stewart等,solid-phase pedtide synthesis,j.am.chem.soc.85:2149-2154,1963)。在体外合成蛋白质可以用手工或自动进行。例如,可以用applied biosystems的431a型肽合成仪(foster city,ca)来自动合成肽。可以分别化学合成本发明蛋白的各片段,然后用化学方法加以连接以产生全长的分子。
[0018]
本发明将克隆得到的tw2odd5基因构建表达载体,经催化实验证实其具有催化对映-贝壳杉烯酸进一步氧化生成2-羟基对映贝壳杉烯酸的生物学功能。
[0019]
因此,本发明的第五方面,提供tw2odd5,或tw2odd5编码基因tw2odd5、或包含所述
编码基因tw2odd5的表达载体、以及包含所述表达载体的宿主菌,在调节和/或合成二萜类化合物中的应用。
[0020]
本发明中,所述的二萜类化合物优选为贝壳杉烷型二萜类化合物,如2-羟基对映贝壳杉烯酸等。
[0021]
本发明的第六方面,提供了一种组合物,所述组合物包含本发明第一方面所述的酶,以及其它合成对映-贝壳杉烯酸所需要的酶。
[0022]
本发明中,还可将tw2odd5或其基因tw2odd5与其它酶或其基因联合使用,使用的方式,如可以是先后使用,也可以是同时使用。
[0023]
还可以通过如下方式制备本发明所述贝壳杉烷型二萜类化合物,即采用生物合成方法,所述方法包括:将tw2odd5的编码基因tw2odd5导入大肠杆菌得到重组大肠杆菌工程菌,发酵重组大肠杆菌,得到松香烷型二萜类化合物,所述大肠杆菌如为大肠杆菌de3菌株。
[0024]
本发明提供的贝壳杉烷型二萜类化合物的生物合成方法,将tw2odd5基因表达盒整合到大肠杆菌中,通过发酵产生α-酮戊二酸依赖型双加氧酶,并以对映贝壳杉烯酸为底物,催化生产2-羟基对映贝壳杉烯酸。本发明对于培育高品质的药用植物品种特别是培育具有高2-羟基对映贝壳杉烯酸含量的雷公藤品种具有重要的理论及实际意义。
[0025]
指α-酮戊二酸依赖型双加氧酶
附图说明
[0026]
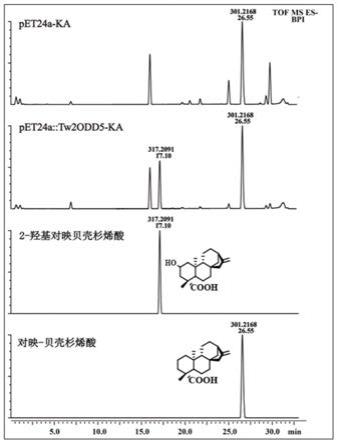
图1.tw2odd5催化产物uplc/q-tof结果图,自上而下依次为空载体产物、阳性载体产物、2-羟基对映贝壳杉烯酸标准品、对映贝壳杉烯酸标准品。
[0027]
图2.tw2odd5蛋白纯化图,左边条带为marker,右边条带为纯化后的蛋白。
[0028]
图3.twodd5酶促动力学常数测定,a为2-羟基对映贝壳杉烯酸产物定量标准曲线;b为tw2odd5酶动力学曲线:一定范围内酶促反应速度随底物浓度升高达到200mm后速度趋于稳定。
具体实施方式
[0029]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0030]
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0031]
下述实施例中的定量试验,均设置三次重复实验,结果取平均值。
[0032]
下述实施例中的雷公藤(tripterygium wilfordiihook.f.)悬浮细胞在文献“王家典,赵瑜君,张逸风,et al.twhmgr过表达对雷公藤甲素和雷公藤红素生物合成的影响[j].药学学报,2018,v.53(08):37-44.”中公开过,公众可从首都医科大学分子生药与中药资源实验室获得。
[0033]
下述实施例中的对映-贝壳杉烯酸(ent-kauranoic acid)是成都普思公司产品,cas号为6730-83-2。
[0034]
gene jet gel extraction kit试剂盒,e.z.n.atm plasmid mini kit i试剂盒购自美国omega公司,peasy-uni seamless cloning and assembly kit试剂盒,2
×
easytaq pcr supermix试剂盒,peasy-blunt zerocloning vector试剂盒,6
×
dnaloading buffer,transetta(de3)感受态细胞,trans1-t1感受态细胞,blueprotein marker,6
×
protein loading buffer是北京全式金生物技术有限公司的产品;phusion high-fidelity pcr master mix with hf buffer和各种核酸内切酶均购自美国neb公司;植物总rna提取试剂盒购自上海普洛麦格生物产品有限公司;fast quant cdna第一链合成试剂盒购自北京天根生化科技有限公司。
[0035]
pet24a质粒购自北京新跃时代公司,该载体通过bamhi、noti内切酶双酶切后连接雷公藤tw2odd5基因,命名为pet24a::tw2odd5,保存于首都医科大学中药资源学与分子生药学实验室,具体构建方法见参考文献(tu l,su p,zhang z,et al.genome of tripterygium wilfordii and identification of cytochrome p450 involved in triptolide biosynthesis[j].nature communications,2020,11(1).)
[0036]
实施例1、雷公藤tw2odd5全长cdna序列的克隆
[0037]
1.雷公藤悬浮细胞总rna提取及cdna第一链的获得
[0038]
使用植物总rna提取试剂盒,按照说明书进行雷公藤悬浮细胞的总rna提取。使用fast quantcdna第一链合成试剂盒,按说明书将总rna反转为cdna。
[0039]
2.引物设计
[0040]
根据雷公藤转录组数据注释筛选得到基因orf序列片段,设计tw2odd5-f和tw2odd5-r引物,引物序列如下:
[0041]
tw2odd5-f:atggacccaccattacaagag
[0042]
tw2odd5-r:ttacacaacaaaccttttgag
[0043]
3.pcr扩增
[0044]
dna聚合酶采用高保真dna聚合酶(phusion high-fidelity pcr master mix)。
[0045]
以步骤1获得的cdna为模板,以tw2odd5-f和tw2odd5-r为引物,采用phusiondna高保真酶进行pcr扩增,得到pcr扩增产物。
[0046]
pcr反应程序:98℃预变性30s;98℃ 10s,60℃ 15s,72℃ 2min,35个循环;72℃延伸5min。
[0047]
取pcr产物与6
×
dnaloading buffer预混合,在1.5%琼脂糖凝胶上以低电压(约5vcm-1
)电泳30-60min;用解剖刀或剃刀片切割含有dna片段的凝胶,尽可能的靠近dna片段切割,以减小凝胶的含量,将胶片放在事先称重的1.5ml离心管并称重。将凝胶按照gene jet gel extraction kit琼脂糖凝胶回收试剂盒按照说明书操作回收。
[0048]
4.克隆载体链接
[0049]
使用peasy-bluntzero cloning vector试剂盒,按照说明书将全长克隆得到的胶回收产物与克隆载体连接,转化至trans1-t1感受态细胞中,培养、鉴定阳性克隆并测序。
[0050]
测序结果表明:pcr扩增产物的序列如序列1所示,将序列1所示的基因命名为tw2odd5,其编码由331个氨基酸残基组成的蛋白质,该蛋白命名为tw2odd5,该蛋白的氨基酸序列为序列2。该克隆载体命名为peasy-blunt-tw2odd5质粒,存于-20℃冰箱。
[0051]
实施例2、雷公藤tw2odd5生物学功能研究
[0052]
1.原核表达载体构建
[0053]
(1)线性化空载体制备:
[0054]
采用neb公司限制性内切酶bamhi、noti对实验室留存的pet24a空载体进行双酶切,切胶回收酶切产物;
[0055]
(2)pcr产物(目的基因)的制备:
[0056]
以含有雷公藤tw2odd5基因全长cdna的载体peasy-blunt-tw2odd5质粒为模板,在引物5’端加15-25bp载体同源臂序列(下划线部分),采用phusiondna高保真酶进行pcr扩增基因编码区。pcr程序:98℃30s,1循环;98℃10s,60℃10s,72℃2min 30s,35循环;72℃5min;4℃维持。
[0057]
tw2odd5-24abamhi-f:agcatgactggtggacagcaaatgggtcgcatggacccaccattac
[0058]
tw2odd5-24anoti-r:tggtggtggtggtgctcgagtgcggccacaacaaaccttttg
[0059]
(3)胶回收线性载体及片段:
[0060]
取pcr产物与6
×
dnaloading buffer预混合,在1.5%琼脂糖凝胶上以低电压(约5vcm-1
)电泳30-60min;用解剖刀或剃刀片切割含有dna片段的凝胶,尽可能的靠近dna片段切割,以减小凝胶的含量,将胶片放在事先称重的1.5ml离心管并称重。将凝胶按照gene jet gel extraction kit琼脂糖凝胶回收试剂盒按照说明书操作回收。
[0061]
(4)表达载体连接:
[0062]
使用peasy-uni seamless cloning and assembly kit试剂盒,按照说明书将线性化载体与pcr产物的轻轻混合,50℃,20min反应,体系如下:
[0063][0064]
线性化空载体与目的基因片段的摩尔比为1:2,其中线性化空载体0.01-0.02pmols,pmols=ng/(片段长度bp
×
0.65kda);
[0065]
连接产物经转化、阳性克隆的初步筛选,送样测序鉴定,得到经测序核苷酸序列无突变重组质粒pet24a::tw2odd5。
[0066]
2.诱导目的蛋白表达
[0067]
(1)将已转化入表达载体的pet24a::tw2odd5阳性单克隆菌落(大肠杆菌de3菌株),于37℃和250r/min的摇床中过夜培养,再按百分之一的比例扩大培养到200ml含50mg/ml kana的液体培养基中,37℃摇培到od600约为0.6-0.8后,加入异丙基硫代半乳糖苷(iptg)至终浓度为0.6mm,低温16℃诱导表达16h;
[0068]
(2)低温诱导的菌液在4℃8000g条件下离心3min收集菌体,加入5ml resuspension buffer(50mm tris-hcl ph8.0,500mm nacl)重悬菌体,超声破碎5min(超声5s,暂停5s),4℃12000g离心10min,取上清作为粗酶反应。
[0069]
3.酶促反应
[0070]
根据参考文献报道(zhang y,su p,wu x,et al.the gibberellin 13-oxidase that specifically converts gibberellin a9 to a20 in tripterygium wilfordii isa 2-oxoglutarate-dependent dioxygenase[j].planta,2019.),使用的体外酶促体系如下:
[0071][0072]
上述体系振荡混匀,30℃,100rpm诱导培养3h。
[0073]
4.酶促产物uplc/q-tof ms检测
[0074]
酶促反应体系用等体积乙酸乙酯萃取3次,合并萃取液,氮气吹干后,加入100μl甲醇混匀后,使用uplc/q-tofms检测。uplc/q-tofms液相条件:使用waters acquity uplc hss t3分析色谱柱,流动相为0.1%甲酸水(a),乙腈(b),流速0.4ml/min。0min-5%b,2min-15%b,3min-15%b,30min-90%b。uplc/q-tofms质谱条件:负离子模式,高能扫描时,斜坡碰撞能量设置为20~40ev。数据采集ms范围为50-1500da。
[0075]
酶促结果如下图1所示,图1结果表明,空白载体中没有产物2-羟基对映贝壳杉烯酸产物,而阳性载体中含有该产物,表明tw2odd5具有将底物对映贝壳杉烯酸氧化成2-羟基对映贝壳杉烯酸的作用。
[0076]
实施例3、tw2odd5产物制备及核磁结构鉴定
[0077]
1.产物制备反应
[0078]
(1)取阳性菌株接种到100ml的lb液体培养基(含有50mg/ml kana)中,37℃,250r/min培养过夜;
[0079]
(2)按1%的比例各扩大培养至8l,37℃250r/min培养至od600约为0.6-0.8后,加入iptg至终浓度为0.6mm,低温16℃诱导表达16h;
[0080]
(3)低温诱导的菌液在高速离心机中离心3min(4℃,10000g)收集菌体,弃去培养基。蒸馏水清洗后重悬于200ml resuspension buffer(20mm tris
·
hcl,500mm nacl,ph8.0)中;
[0081]
(4)利用ats机高压均质机,1200psi低温破碎7min后,12000g,低温离心15min,收集上清液;
[0082]
(5)200ml上清液中等比例加入抗坏血酸钠、α-酮戊二酸钠、feso4
·
7h2o和底物对映-贝壳杉烯酸,30℃100rpm/min反应12h。
[0083]
(6)反应液用等体积的乙酸乙酯萃取3次,合并上清液后旋干,用10ml甲醇溶解产物。过0.22μm滤膜后,进样制备液相得到反应产物。
[0084]
(7)制备液相色谱条件
[0085]
使用安捷伦制备液相(agilent 1260)进行分离,色谱柱使用ymc-triart c18(250
×
10mm,5μm)半制备柱,流动相为水(a),乙腈(b),流速5ml/min。0min-5%b,2min-30%b,35min-100%b,收集目标馏分并进行浓缩得到制备产物。
[0086]
(8)将制备得到的反应产物溶解于氘代丙酮至终浓度为6mg/ml,利用800m核磁检测的1h nmr、
13
c nmr谱图。
[0087]1h-nmr谱图(c3d6o-d4,800mhz)结果如下:δ0.72(1h,t,j=11.9hz),0.95(1h,t,j=11.9hz),1.00(3h,s),1.09(1h,m),1.14(1h,d,j=3.7hz),1.14(1h,m),1.26(3h,s),1.50(1h,m),1.54(1h,m),1.66(1h,m),1.66(2h,m),1.87(2h,m),2.02(1h,m),2.09(1h,m),
2.20(1h,m),2.41(1h,m),2.63(1h,m),4.08(1h,m),4.74(1h,s),4.81(1h,s).
[0088]
13
c-nmr谱图(c3d6o-d4,800mhz)结果如下:δ16.4,18.3,21.6,28.4,32.9,39.5,40.7,41.1,43.7,44,44.2,47.1,48.7,49.9,54.9,56.1,62.9,102.8,155.3,178.
[0089]
实施例4、tw2odd5蛋白纯化及酶动力学参数测定
[0090]
1.tw2odd5蛋白纯化
[0091]
(1)发酵2l阳性菌株,具体操作同实施例2、3,诱导蛋白后将得到的粗酶上清液过ni柱(ni柱前端接流动泵,后端接紫外检测仪),3ml/min,重复上样3次,保证蛋白与ni的充分结合;
[0092]
(2)配制含不同浓度咪唑及500mmnacl的tris-hcl缓冲液(ph8.0),按照咪唑浓度从低到高的顺序分别洗脱ni柱后,不同浓度咪唑的洗脱液跑sds-page蛋白胶,纯化得到目的蛋白如图2所示,显示纯化后的蛋白为一分子量约为40kda的条带,即目的蛋白tw2odd5。
[0093]
2.twodd5酶促动力学常数测定
[0094]
(1)将纯化得到的蛋白使用resuspension buffer浓缩后,使用nanodropone测定蛋白浓度,为8mg/ml;
[0095]
(2)使用不同浓度的底物在以下体系反应:
[0096][0097]
25℃反应2h后加入等体积冰甲醇终止反应后,使用uplc/qqq ms进行产物检测,配制标准曲线后进行定量。uplc/qqq ms液相条件:使用waters acquity uplc hss t3分析色谱柱,流动相为0.1%甲酸水(a),乙腈(b),流速0.4ml/min。0min-50%b,2min-50%b,5min-90%b。uplc/q-tofms质谱条件:负离子模式,母离子设为317.00,子离子设为255.10,271.10。定量后处理结果展示如图3所示,图3结果表明,一定范围内线性关系良好,酶促反应速度随底物浓度升高,当底物浓度达到200nm后趋于稳定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1