一种Tubulysin及其类似物的关键中间体的合成方法与流程
一种tubulysin及其类似物的关键中间体的合成方法
技术领域
1.本发明属于医药技术领域,具体涉及一类适用于靶向治疗的细胞毒素分子tubulysin及其类似物的关键中间体的合成方法。
背景技术:
2.tuv片段(化合物a)是合成天然抗癌药物tubulysin家族化合物(结构式如下)的关键中间体,tubulysin及其类似物与tuv片段(化合物a)结构如下:
[0003][0004]
tubulysins不仅具有很高的抗癌活性,而且能够有效地抑制耐药性癌细胞的生长,但它的具体作用机理却是与埃博霉素和紫杉醇相反,即促进微管蛋白的聚合。另外,还发现tubulysins具有抑制血管新生的作用。这些都说明了tubulysins作为抗癌药的巨大潜力,tubulysins不仅是结合到小分子药物缀合物(smdc)递送系统中的理想候选者,而且tubulysins作为adc有效载荷,已经被充分关注和研究。
[0005]
cn110684044a公开了一种制备tubulysin关键片段tuv的方法,反应式如下:
[0006][0007]
p表示氮原子的保护基,具体为boc、cbz,coome或cooet,p1表示氧原子保护基,具体为tbs、tes、tbdps、bn、pmb、ac,r、r1和r2表示不同芳基或烷基取代基。
[0008]
cn104072578b公开了天然产物tubulysin u的制备方法,其中tuv片段的制备方法如下:
[0009][0010]
cn110684044a和cn104072578b公开tuv的制备方法,均存在路线步骤多,且使用了多种危险试剂,没有成本优势,不适合工业化生产。
[0011]
wo2017134547a1公开以下路线中,化合物a作为关键中间体:
[0012][0013]
该路线具有明显优势,避免使用叠氮,钠氢,高碘酸等危险试剂;化合物a是该路线关键中间体,整体收率也较高,但是也没有成本优势。wo2017134547a1、wo2017054080和j.org.chem.2008,73,4362
–
4369也报道了类似方法:
[0014][0015]
目前文献报道包括化合物1先与多聚甲醛环合生成中间体2,然后与氰基硼氢化钠还原,脱保护生成化合物3;根据文献报道类似物的路线,原料纯化难度大,中间体不稳定;第一步环合反应很慢,需要很长时间,且需要密罐反应;还原使用到氰基硼氢化钠和盐酸,有释放剧毒氰化氢的风险;该方案使用范围有限,仅能制备n-甲基化合物。
[0016]
综上所述,化合物a的合成具有明显缺陷,因此,寻找较优的合成路线具有重要意义,需要开发更为合适的合成方法。
技术实现要素:
[0017]
基于背景技术存在的技术问题,化合物a的工艺路线具有迫切的需要和广阔的前景。本发明提出了一种tubulysin及其类似物的关键中间体(化合物a)的合成方法,该合成方法反应条件温和、环保,路线新颖、后处理和纯化简单,制备得到的化合物a纯度好、收率高、成本低、易于放大生产。
[0018]
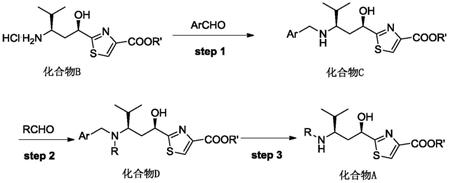
本发明提出的化合物a的一种合成工艺,合成路线如下:
[0019][0020]
r选自c1~c6的脂肪烷基,优选甲基、乙基,丙基,异丙基或丁基,最优选甲基;
[0021]
r’选自甲基或乙基;
[0022]
ar选自苯基或取代苯基,取代苯基选自对甲氧基苯基,对硝基苯甲基,2,4-二甲氧基苯甲基等;
[0023]
包括以下步骤:
[0024]
s1:以化合物b为原料,与archo经过还原胺化反应得到化合物c;
[0025]
s2:再与rcho经还原胺化反应得到化合物d;
[0026]
s3:化合物d经过脱保护反应得到化合物a。
[0027]
本发明进一步的方案:步骤s1的反应溶剂选自dcm,thf,乙腈,甲苯或甲醇,优选dcm或thf;
[0028]
本发明进一步的方案:步骤s1的archo选自苯甲醛,对甲氧基苯甲醛,对硝基苯甲醛,2,4-二甲氧基苯甲醛等,优选苯甲醛;
[0029]
本发明进一步的方案:步骤s1的还原胺化使用还原剂选自nabh(oac)3,nabh4,
nabh3cn,et3sih等,优选nabh(oac)3;
[0030]
本发明进一步的方案:步骤s1中化合物b使用盐酸盐,可选择性碱(例如三乙胺)游离后,再与archo经过还原胺化反应或者同时加入碱(例如三乙胺)和还原剂一锅反应;
[0031]
本发明进一步的方案:步骤s1在反应温度0~50℃进行,优选20~30℃。
[0032]
本发明进一步的方案:步骤s2反应溶剂选自dcm,thf,乙腈,甲苯或甲醇,优选dcm或thf;
[0033]
本发明进一步的方案:步骤s2中rcho选自甲醛,乙醛,丙醛,丁醛等;
[0034]
本发明进一步的方案:步骤s2的还原胺化使用还原剂选自nabh(oac)3,nabh4,nabh3cn,et3sih等,优选nabh(oac)3;
[0035]
本发明进一步的方案:步骤s2在反应温度0~50℃进行,优选20~30℃。
[0036]
本发明进一步的方案:在步骤s3脱保护反应中,当ar选自苯基,s3脱保护反应选自氢化脱保护方案:当ar选自取代苯基,取代苯基选自对甲氧基苯基,对硝基苯甲基,2,4-二甲氧基苯甲基时,s3脱保护反应选自氧化脱保护反应或酸脱保护反应;
[0037]
本发明优选的技术方案,如下:
[0038]
当ar选自苯基,s3脱保护反应选自方法一氢化脱保护方案:
[0039]
方法一,氢化脱保护方案:
[0040]
本发明进一步的方案:步骤s3中氢化反应脱保护在甲醇,乙醇,thf,乙腈等溶剂中进行,优选甲醇;
[0041]
本发明进一步的方案:步骤s3中氢化反应脱保护在催化剂和氢源作用下反应;
[0042]
本发明进一步的方案:步骤s3中氢化反应脱保护的催化剂选自钯碳,铂炭,raney ni等,优选钯碳;
[0043]
本发明进一步的方案:步骤s3中氢化反应脱保护的氢源选自氢气,甲酸铵,甲酸或环己二烯,优选氢气;
[0044]
本发明进一步的方案:步骤s3中氢化反应脱保护在反应温度0~50℃进行,优选20~30℃;
[0045]
当ar选自取代苯基,s3脱保护反应选自方法二氧化脱保护方案或方法三s3酸脱保护方案:
[0046]
方法二,s3氧化脱保护方案:
[0047]
本发明进一步的方案:步骤s3中氧化脱保护在乙腈,乙醇,thf,水等,优选乙腈;
[0048]
本发明进一步的方案:步骤s3中氧化脱保护在氧化剂作用下反应;
[0049]
本发明进一步的方案:步骤s3中氧化脱保护的氧化剂选自过硫酸钾,ddq,双氧水等,优选过硫酸钾;
[0050]
本发明进一步的方案:步骤s3中氧化脱保护在反应温度20~100℃进行,优选65~70℃;
[0051]
方法三,s3酸脱保护方案:
[0052]
本发明进一步的方案:步骤s3中酸脱保护的酸选自三氟乙酸,硫酸,甲磺酸等,优选三氟乙酸;
[0053]
本发明进一步的方案:步骤s3中酸脱保护在反应温度60~120℃进行,优选70~80℃;
[0054]
本发明进一步的方案:步骤s3中在无溶剂体系下反应;
[0055]
本发明进一步的方案:步骤s3中酸脱保护酸的酸体积用量为化合物d(原料)体积用量的1~20倍,优选10~15倍。
[0056]
本发明还提供一种新的中间体化合物:
[0057][0058]
r’选自甲基或乙基;
[0059]
r1选自苯基或取代苯基,取代苯基选自对甲氧基苯基,对硝基苯基,2,4-二甲氧基苯甲基,优选苯基;r2选自对甲氧基苯基,对硝基苯基或2,4-二甲氧基苯甲基。
[0060]
本发明进一步的方案,还提供一种新的中间体化合物:
[0061][0062]
本发明还提供一种制备tubulysin及其类似物的方法,包含以上所述的合成方法或所述的中间体化合物。
[0063]
本发明有益的技术效果:
[0064]
本发明直接采用易纯化的化合物b做起始物料,化合物b为盐酸盐且固体形态,易于操作;化合物b经过还原胺化反应制备二级胺中间体,再经另一还原胺化反应得到新的三级胺中间体,三级胺中间体选择性经过氢化脱保护或氧化脱保护或酸脱保护得到新产品。整个反应过程不使用叠氮化合物等危险试剂,也无剧烈条件,纯化效果较好,简便快捷,反应条件温和可控,三步收率各步均达到80%以上,总收率高,成本优势明显,易于工业化生产;
[0065]
特别是现有技术没有关于对化合物a上n甲基单取代的制备方法报道,本发明通过巧妙地设计,提供了一种单烷基甲基取代的方法;该方法利用苄基或任意取代苄基保护氨基,然后与甲醛还原胺化反应后,脱除苄基或任意取代苄基保护后得到单烷基甲基的中间体;实现了该领域的研究空白,反应收率高、产品质量好,避免了双烷基化杂质;反应具有良好选择性,同时解决了现有技术反应选择性差的问题。c19h26
n2o3s[m+h]+:计算值363.2,实测值363.1),无需纯化直接进入下一步;
[0082]
上步粗品加入30ml thf和3ml 37%甲醛水溶液在20-30℃搅拌3小时,tlc无原料,加入5.1g醋酸硼氢化钠,搅拌18小时中控反应完成;反应体系加入饱和碳酸氢钠水溶液至ph为7-8,静置分层,加入30ml乙酸乙酯萃取水相;有机相合并,用9ml饱和食盐水洗涤一次,有机相浓缩至干,粗品柱层析(dcm/meoh=20:1洗脱),即得化合物2d:2.9g无色油状物,两步收率:79.2%。1h nmr(400mhz,d6-dmso)δ8.44(s,1h),7.36-7.22(m,5h),6.60(brs,1h),5.14-5.12(m,1h),4.33-4.27(m,2h),3.81-3.78(dd,j=12.0hz,1h),3.68-3.64(dd,j=16.0hz,1h),2.76-2.71(m,1h),2.18(s,3h),2.12-2.05(m,1h),1.97-1.92(m,1h),1.71-1.64(m,1h),1.32-1.29(t,j=8.0hz,3h),0.97-0.96(d,j=4.0hz,3h),0.92-0.90(d,j=8.0hz,3h)。esi m/z c
20h28
n2o3s[m+h]
+
:计算值437.2,实测值437.2。
[0083]
实施例3:
[0084][0085]
将1.0g化合物b溶于10ml四氢呋喃中,加入0.4g三乙胺和0.57g 2,4-二甲氧基苯甲醛,保持室温搅拌1小时;然后分批加入1.7g醋酸硼氢化钠,继续室温搅拌3小时后,tlc监控反应完成;
[0086]
反应体系滴入饱和碳酸氢钠水溶液至ph为7-8,静置分层,加入10ml乙酸乙酯萃取水相;有机相合并,用3ml饱和食盐水洗涤一次,有机相浓缩至干即得化合物3c(esi m/z c
21h30
n2o5s[m+h]
+
:计算值423.2,实测值423.1),无需纯化直接进入下一步;
[0087]
上步粗品加入10ml thf和1ml 37%甲醛水溶液在20-30℃搅拌3小时,tlc无原料,加入1.7g醋酸硼氢化钠,搅拌18小时中控反应完成;反应体系加入饱和碳酸氢钠水溶液至ph为7-8,静置分层,加入10ml乙酸乙酯萃取水相;有机相合并,用3ml饱和食盐水洗涤一次,有机相浓缩至干,粗品柱层析(dcm/meoh=20:1洗脱),即得化合物3d:1.2g无色油状物;两步收率:85.1%。1h nmr(400mhz,cdcl3)δ8.02(s,1h),7.27(s,1h),7.03-7.01(d,j=8.0hz,1h),6.46(brs,1h),6.40-6.38(d,j=8.0hz,1h),5.24-5.22(m,1h),4.61-4.38(m,2h),3.84(s,3h),3.81(s,3h),3.71-3.68(dd,j=12.0hz,1h),3.50-3.47(dd,j=12.0hz,1h),2.55-2.53(m,1h),2.30(s,3h),2.31-2.27(m,1h),2.12-2.07(m,2h),1.41-1.37(t,j=8.0hz,3h),0.93-0.91(d,j=8.0hz,3h),0.79-0.78(d,j=4.0hz,3h)。esi m/z c
22h32
n2o5s[m+h]+:计算值437.2,实测值437.2。
[0088]
实施例4:
[0089]
化合物a的合成:
[0090][0091]
r选自c1~c6的脂肪烷基,优选甲基、乙基,丙基,异丙基或丁基,最优选甲基;
[0092]
r’选自甲基或乙基;
[0093]
ar选自苯基或取代苯基,取代苯基选自对甲氧基苯基,对硝基苯甲基,2,4-二甲氧
基苯甲基等。
[0094]
氢化反应脱bn为例:
[0095][0096]
将1.0g化合物2d溶于10ml乙醇中,加入0.3g 10%pd/c,置换氢气;室温搅拌反应,待中控完成;过滤,滤液浓缩至干,柱层析(dcm/meoh=10:1洗脱)即得淡黄色色固体化合物a:0.7g,收率:91%。1h nmr(400mhz,cd3od)δ8.38(s,1h),5.22-5.19(m,1h),4.44-4.39(m,2h),2.83-2.81(m,1h),2.60(s,3h),2.13-2.08(m,3h),1.44-1.40(t,3h),0.99-0.98(d,6h)。
[0097]
氧化反应脱dmb为例:
[0098][0099]
将1.0g化合物3d溶于20ml乙腈/水(1:1体积比)中,加入1.24g过硫酸钾,升温至65-70度搅拌反应2小时,中控转化完全;降至室温,反应液浓缩,加入乙酸乙酯萃取,有机相浓缩至干,柱层析(dcm/meoh=10:1洗脱)即得淡黄色固体化合物a:0.5g,收率:76%。
[0100]
tfa酸脱dmb为例:
[0101][0102]
将2.0g化合物3d溶于20ml三氟乙酸中,升温至回流搅拌50小时,中控反应完成;降至室温,反应液浓缩,加入10ml乙酸乙酯,用饱和碳酸氢钠水溶液调ph至7-8,静置,分层;水相再用10ml乙酸乙酯萃取一次次,合并有机相后加入无水硫酸钠干燥,过滤,母液浓缩至干,经柱层析分离(dcm/meoh=10:1洗脱),即得淡黄色色固体化合物a:1.1g,收率:84%。
[0103]
对比实施例:
[0104][0105]
将1.0g化合物b加入10ml thf和1ml 37%甲醛水溶液在20-30℃搅拌3小时,然后加入1.7g醋酸硼氢化钠,搅拌18小时中控反应,hplc数据显示化合物b:化合物a:化合物e=12.7/19.0/66.1。
[0106]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此;
[0107]
任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术
方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1