远志呫吨酮Ⅲ的对照品的制备方法及固体对照品与流程
对照品。
23.在本发明优选的实施方式之中,所述含有远志呫吨酮ⅲ的原料提取物粗品的制备方法为将原料远志药材粉碎,利用醇系的水溶液浸泡,并在65~80℃进行回流提取,过滤得滤液,将滤液在30~80℃的温度下避光浓缩,得到远志呫吨酮ⅲ的原料提取物粗品。
24.在本发明优选的实施方式之中,正相色谱纯化工序中,所述酯类溶剂:醇类溶剂的比例为8~1:1。
25.在本发明优选的实施方式之中,聚酰胺色谱纯化工序中,所述酯类溶剂:醇类溶剂的比例为4~1:1。
26.在本发明优选的实施方式之中,所述酯类溶剂为乙酸乙酯或乙酸丁酯,所述醇类溶剂为甲醇或乙醇。
27.在本发明优选的实施方式之中,中压反相色谱纯化工序中,用浓度5~50%的乙腈水溶液以压力为0.1~0.3mpa进行洗脱。
28.在本发明优选的实施方式之中,醇系的水溶液为50~70%的甲醇水溶液或者50~70%的乙醇水溶液。
29.在本发明优选的实施方式之中,正相色谱纯化工序中,正相色谱柱的填料为粒径为100~300目的硅胶,中压反相色谱柱的填料为粒径为40~60μm的十八烷基硅烷键合硅胶。
30.在本发明优选的实施方式之中,溶剂处理工序中:用以质量比计5倍~20倍的水将远志呫吨酮ⅲ半成品溶解,在第1滤液中,缓慢加入的甲醇的量为滤液体积的0.2~0.5倍。
31.本发明的另一方面,还提供一种纯度为98.5%以上的远志呫吨酮ⅲ的固体对照品,其通过上述本发明的制备方法制备得到。
32.与现有技术相比,本发明具有如下特点:
33.1、本专利能够获得大量的远志呫吨酮ⅲ纯品,工艺成熟稳定,为远志药材的质量控制提供坚实的标准物质原料,具有较高的经济价值。
34.2、本发明分离过程中采用利用了苯乙烯类-二乙烯苯骨架大孔树脂吸附性与正相色谱层析、聚酰胺的协同效应,对远志呫吨酮ⅲ的hplc纯度有显著的提升,本发明还利用了远志呫吨酮ⅲ在不同溶剂间的溶解度差异,进一步提高了纯度,这些操作都简单易行,容易工业化放大。本发明通过溶剂法处理处理得到98.5%以上纯品,试剂用量少、效率高、成本低、更加环保、利于规模化生产。
附图说明
35.图1为本发明实施例1的远志呫吨酮ⅲ提取大致的流程图;
36.图2为本发明实施例1获得的远志呫吨酮ⅲ纯品的ms图谱;
37.图3为远志呫吨酮ⅲ纯品的h-nmr图谱(dmso-d6,600mhz);
38.图4为远志呫吨酮ⅲ纯品的c-nmr图谱(dmso-d6,150mhz);
39.图5为实施例1获得远志呫吨酮ⅲ纯品的hplc谱图;
40.图6为比较例1获得远志呫吨酮ⅲ纯品的hplc谱图。
具体实施方式
41.以下记载本发明的具体实施方式。
42.本发明中,远志呫吨酮ⅲ有时也称为目标物、或分离目标物。
43.如果并未特殊说明,本发明中目标物的纯度和含量的百分数,为以质量比计的含量。如果并未特殊说明,本发明中液体与液体含量配比的百分数,例如甲酸在乙腈水溶液中的含量配比,以体积分数计量。
44.本发明通过简单高效的操作流程,能够分离得到纯度很高的远志呫吨酮ⅲ。本发明的制备分离方法依次包括以下步骤:
45.依次包括以下步骤:
46.富集浓缩工序:将含有远志呫吨酮ⅲ的原料提取物粗品水溶液上样至苯乙烯类-二乙烯苯骨架大孔树脂吸附柱,用大孔树脂吸附柱体积10~30倍的水洗脱去除杂质,再利用大孔树脂吸附柱体积3~10倍的以体积比计10~25%的甲醇水溶液或者10~25%的乙醇水溶液洗脱去除杂质,再利用大孔树脂吸附柱体积3~10倍的以体积比计30~60%的甲醇水溶液或者30~60%的乙醇水溶液洗脱获得含目标物的溶液,浓缩得到含有远志呫吨酮ⅲ的第一粗品的工序;
47.正相色谱纯化工序:将含有远志呫吨酮ⅲ的第一粗品上样至正相色谱柱,用包含酯类溶剂、醇类溶剂、水的混合洗脱液进行洗脱,酯类溶剂:醇类溶剂的比例为10~0.8:1,醇类溶剂:水的比例为5~2:1根据tlc或者hplc检测收集洗脱液,将洗脱液浓缩、干燥,得到第二粗品;
48.聚酰胺色谱纯化工序:将含有第二粗品上样至聚酰胺色谱柱,用包含酯类溶剂、醇类溶剂、水的混合洗脱液进行洗,酯类溶剂:醇类溶剂的比例为8~3:1,醇类溶剂:水的比例为8~3:1根据tlc或者hplc检测收集洗脱液,将洗脱液浓缩、干燥,得到第三粗品;
49.中压反相色谱纯化工序:将第三粗品上样至中压反相色谱,用浓度5~60%的乙腈水溶液或者甲醇水溶液以压力为0.02~1mpa进行洗脱,根据tlc或者hplc检测收集含目标物的洗脱液,浓缩、去除溶剂,得到远志呫吨酮ⅲ半成品;
50.溶剂处理工序:将远志呫吨酮ⅲ半成品利用水进行溶解,过滤得到第1滤液,在第1滤液中,缓慢加入甲醇或者乙醇,析出固体沉淀,过滤沉淀,干燥,得到远志呫吨酮ⅲ对照品。
51.本发明的发明人发现,常规的逐渐增加色谱分离的梯度的分离方法,无法得到大量得到纯度很高的远志呫吨酮ⅲ,推测原因可能是远志呫吨酮ⅲ因有羟基含量太多,在正相色谱上的吸附过于严重,因此难以利用正相色谱的梯度洗脱效果,为此发明人不断尝试各类有效的纯化分离方法的组合,降低正相色谱在整个分离纯化中的应用比例,通过不断探索,发现苯乙烯类-二乙烯苯骨架大孔树脂吸附性与正相色谱层析、聚酰胺的协同使用,可以获得非常好的纯化效果和收率。本发明的方法中最终步骤的溶剂处理工序,也对于提高纯度起到至关重要的作用,该步骤中特定的溶剂的配合,使得前期步骤中不能分离的杂质部分被大部分去除。上述工序的顺序很重要,不能轻易替换。本发明的分离方法一个特别之处在于色谱柱分离工序和大孔树脂吸附柱的有机组合和协同,利用大孔树脂的吸附作用,迅速提高目标物纯度,是本发明首次使用的方法。具体进行以下说明。
52.富集和浓缩工序中利用苯乙烯-二乙烯苯骨架大孔树脂对于远志呫吨酮ⅲ的特异
性吸附作用,可以迅速的提高目标物的纯度,具体原因尚不清楚,可能是由于苯乙烯-二乙烯苯骨架的大孔树脂的特定结构对于远志呫吨酮ⅲ特异性更强,发明人也尝试过苯乙烯-丙烯酸酯类的大孔树脂,发现收率上会比苯乙烯-二乙烯苯骨架大孔树脂差。因此在富集和浓缩步骤中,优选使用苯乙烯-二乙烯苯骨架,作为常用的苯乙烯-二乙烯苯骨架大孔树脂,可以使用ab-8、hpd-100、d-101等型号的大孔树脂等。苯乙烯-二乙烯苯骨架大孔树脂的孔径、粒度等没有特别的限制,从吸附的效果来看,优选使用孔径为5nm~100nm的大孔树脂。在富集和浓缩工序中,优选利用35~45%的甲醇水溶液或者35~45%的乙醇水溶液洗脱获得含目标物的溶液,从环境毒性和分离效果平衡的角度出发,更优选使用38%~42%的乙醇水洗脱获得目标物溶液,特别优选使用40%的乙醇水洗脱。富集工序中的,利用10~25%的甲醇水溶液或者10~25%的乙醇水溶液洗脱去除杂质的工序非常重要,能极大提高本发明整体的分离效率和回收率,进一步优选利用20%~25%的乙醇水洗脱去除杂质。
53.本发明中过滤的方法没有特别限制,可以使用常规的过滤器、抽滤装置、微孔滤膜装置进行过滤。本发明中的干燥也没有特别限制,可以使用烘箱、红外灯、加热板等,干燥的温度,本领域人员可以适当选择。本发明中,浓缩方法以及除去有机溶剂的方法,没有特别限制,可以使用加热蒸发浓缩,旋转蒸发仪浓缩等。本发明中使用的水,没有特别限制,可以使用自来水、蒸馏水、去离子水等常用的水。
54.上述的正相色谱纯化工序的目的是将极性差别较大的杂质与目标物区分开,正相色谱纯化工序中,原料粗品上样的方式并无特别限制,可以采用良溶剂溶解滴加上样,也可以通过填料拌样上样,为了将上样体积压缩的较低,优选拌样填料上样。
55.正相色谱纯化工序所使用的正相色谱柱的填料,可以用公知的正相填料,所谓正相填料,就是固定相的极性大于流动相的极性的填料,如果流动相为有机溶剂,常用的填料为硅胶(具体为sio2,二氧化硅)、al2o3、极性键合相填料等,本发明中优选使用硅胶。硅胶的粒径大小没有特别限制,从效率和填料易得的角度出发,在优选的本发明的制备方法中,第一正相色谱柱提纯工序中,正相色谱柱的填料为粒径为100~300目的硅胶。为了更好的获得分离效率,并且从分离度和吸附损失的角度出发,优选使用硅胶,进一步优选使用100~300目的硅胶。
56.本发明中正相色谱的洗脱剂为酯类溶剂、醇类溶剂和水的组合,这是由于本发明的目标物与正相色谱的吸附力较强,因此使用了极性较大的溶剂系统。
57.作为酯类溶剂,合适于本发明方法的酯类溶剂例如为甲酸或乙酸的c2-c8醇酯。可举出的酯类溶剂包括但不限于甲酸丁酯、乙酸丙酯、乙酸异戊酯、乙酸异丁酯、乙酸异丙酯、乙酸正戊酯、乙酸乙酯、乙酸丁酯、乙酸辛酯、乙酸异辛酯、乙酸仲辛酯等。可单独使用一种酯类溶剂或使用两种或更多种酯类溶剂的混合物。本发明中,进一步优选使用乙酸乙酯或乙酸丁酯,更进一步优选使用乙酸乙酯。
58.醇类溶剂,可使用常用的甲醇、乙醇、异丙醇。作为本发明中的优选组合,可以使用乙酸乙酯和甲醇的组合,获得更好的分离效果。
59.在正相色谱纯化工序中,酯类溶剂:醇类溶剂的混合比例优选为8~1:1。
60.本发明中的聚酰胺色谱纯化工序安排在正相色谱和反相色谱之间的顺序非常重要。聚酰胺(polyamide)是通过酰胺基聚合而成的一类高分子化合物,层析分离中常用的聚酰胺是由己内酰胺聚合而成的尼龙6和由己二酸和己二胺聚合而成的尼龙66。聚酰胺分子
中含有丰富的酰胺基团,可与酚类、醌类、硝基化合物等形成氢键而被吸附,与不能形成氢键的化合物分离。吸附的强度主要取决于这两种化合物中羟基的数目与位置、以及溶剂与化合物或溶剂与聚酰胺之间形成氢键的缔合能力大小。溶剂分子与聚酰胺或黄酮类化合物形成氢键缔合的能力越强,则聚酰胺对这两种化合物的吸附作用将越弱。
61.聚酰胺层析柱即是利用此性质对本发明中的目标物以及与目标物类似的化合物等进行吸附、洗脱而分离。酰胺吸附属于氢键吸附,系通过其分子中众多的酰胺羰基与酚类、黄酮类化合物的酚羟基,或酰胺键上的游离胺基与醌类、脂肪羧酸上的羰基形成氢键缔合而产生吸附。因此,聚酰胺吸附色谱特别适合分离本发明的目标物。聚酰胺对被分离物质吸附力的大小取决于被分离物质分子结构中可与聚酰胺形成氢键缔合的基团数目及氢键作用强度。
62.常用的柱层析用聚酰胺粉末为10-400目的颗粒,本发明中可以使用市售的柱层析用聚酰胺粉末装柱,优选使用30~100目的聚酰胺粉末装柱进行分离纯化。
63.本发明的聚酰胺色谱纯化工序中的上样没有特别限制,可以采用拌样上样,或者溶剂溶解上样。洗脱剂对于得到本发明的目标物而言至关重要,本发明中用包含酯类溶剂、醇类溶剂、水的混合洗脱液进行洗,酯类溶剂:醇类溶剂的比例为8~3:1,醇类溶剂:水的比例为8~3:1,其中酯类溶剂:醇类溶剂的实际例子与上述正相色谱纯化工序中举出的例子相同。作为本发明中的优选组合,可以使用乙酸乙酯和甲醇的组合,获得更好的分离效果。酯类溶剂:醇类溶剂的比例进一步优选为4~1:1。
64.本发明中,中压反相色谱分离用于将与目标物极性有差异的其他成分分离。该工序中,上样的方式并无特别限制,可以采用良溶剂溶解滴加上样,也可以通过填料拌样上样,为了将上样体积压缩的较低,优选拌样填料上样。该工序中采用0.02~1mpa的洗脱压力能够实现分辨率和速度之间的平衡,进一步优选的柱压力是0.05~0.5mpa。中压柱分离工序中,中压反相色谱柱的压力更进一步优选为0.1mpa~0.3mpa,此压力因为能平衡分离效果和分离速度而特别优选。本发明的中压反相色谱纯化工序中,进一步优选用浓度5~60%的乙腈水溶液溶液以压力为0.1~0.3mpa进行洗脱。本发明中,为了获得更好的分离效果,优选利用梯度洗脱的方法,例如使用5%、10%、20%、50%乙腈水梯度洗脱。
65.本发明中色谱柱制备工序中使用的反相色谱柱的反相填料,可以用公知的非极性的,键合的官能团为烷烃的(例如:c18(ods)、c8、c4等)的硅胶。优选c18(ods)硅胶柱,即十八烷基硅烷键合硅胶,优选使用40~60μm的填料,合理的粒径有利于维持适当的柱压和分辨率,十八烷基硅烷键合硅胶在市场上易于购买。从效率和填料易得的角度出发,在优选的本发明的制备方法中,反相色谱柱的填料为粒径为40~60μm的十八烷基硅烷键合硅胶。
66.上述溶剂处理工序,安排在最后工序,可以充分利用目标物在水和有机溶剂中溶解度差异来提高纯度。
67.溶剂处理工序,优选用以质量比计50倍~100倍的水溶解远志呫吨酮ⅲ半成品得到第1滤液中,加入甲醇或者乙醇就会使目标物析出,加入甲醇或乙醇的量没有特别限制,本领域人员可以根据实际情况选择,一般而言缓慢加入的为滤液体积的0.2~0.5倍的乙醇或者甲醇,优选0.3~0.5倍。溶剂处理工序中,使用甲醇或乙醇,和水进行溶剂处理是重要的,能够使本发明的效果最大化。
68.本发明中,所谓远志呫吨酮ⅲ半成品即使没有溶剂处理工序,也能够再利用常规
的提纯手段,例如高压柱色谱分离等获得远志呫吨酮ⅲ,然而,为了获得高纯度的远志呫吨酮ⅲ,需要更多的溶剂、时间,同时还会极大的降低回收率,最后的溶剂处理可以极大的提高效率。
69.溶剂处理之前,优选将远志呫吨酮ⅲ半成品研磨成粉末。
70.本发明中,所谓的含有远志呫吨酮ⅲ的原料提取物粗品,可以是任意的含远志呫吨酮ⅲ的提取物,例如市售的远志药材提取物。为了使本发明获得更优异的分离效果和回收率,本发明中,优选含有远志呫吨酮ⅲ的原料提取物粗品的获得方法为:
71.将原料远志药材粉碎,利用醇系的水溶液浸泡,并在65~80℃进行回流提取,过滤得滤液,将滤液在30~80℃的温度下避光浓缩,得到远志呫吨酮ⅲ的原料提取物粗品。
72.此时,在本发明优选的实施方式中,上述的醇系的水溶液为50%~70%的甲醇水溶液或者50%~70%的乙醇水溶液。回流提取可以使用通常的方法,例如使用含有冷凝器的提取器,加热提取液的方法,利用提取罐、索式提取器的方法等。
73.本发明还提供一种纯度为98.5%以上的远志呫吨酮ⅲ的固体对照品,其通过上述制备方法制备得到。
74.基于以上说明可知,本发明具有以下特点:本发明操作简单高效,能够进行工艺放大,分离得到大量远志呫吨酮ⅲ单体。利用了苯乙烯类-二乙烯苯骨架大孔树脂吸附性与正相色谱层析、聚酰胺的协同效应,对远志呫吨酮ⅲ的hplc纯度有显著的提升,本发明还利用了远志呫吨酮ⅲ在不同溶剂间的溶解度差异,进一步提高了纯度,。本发明可以很容易的获得高于98.5%的纯度的远志呫吨酮ⅲ,特别适合作为高质量的对照品使用。同时整个过程适合放大,可以极大的提高对照品的制备通量。
75.实施例
76.以下,通过实施例对本发明进行详细描述,但并不意味着对本发明任何不利限制。本文已经详细地描述了本发明,其中也公开了其具体实施例方式,对本领域的技术人员而言,在不脱离本发明精神和范围的情况下针对本发明具体实施方式进行各种变化和改进将是显而易见的。
77.实施例中所使用的试剂购自aldrich公司、国药试剂公司等公司。仪器条件如下:
78.ms分析条件
79.仪器sciex triple tof 4600lc/ms
80.检测模式negative ion mode
81.esi源参数:
[0082][0083]
hplc色谱条件
[0084]
1.1供试品溶液制备
[0085]
样品1.8mg,加50%甲醇水3ml溶解,配制成0.60mg/ml。
[0086]
1.2hplc色谱条件
[0087][0088]
流动相组成a-水(0.1%甲酸),b-乙腈
[0089][0090]
实施例1远志药材中远志呫吨酮ⅲ对照品的制备方法
[0091]
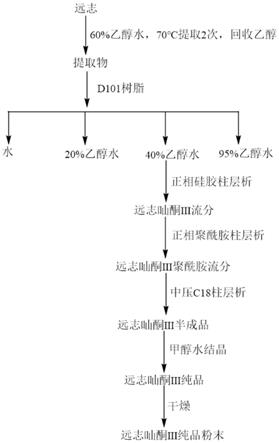
按照图1的流程,按如下工艺步骤进行,远志中远志呫吨酮ⅲ的制备方法步骤如下:
[0092]
a、药材提取:取远志药材100kg,产地四川,粉碎成粗颗粒状,注意不可粉碎过细,否则会导致药材粉末阻力大,不利于溶剂充分扩散,加入60%乙醇水1000l,70℃回流提取3h,提取2次,合并提取液并过滤,注意过滤很重要,否则引入的药渣将影响下一步树脂富集效果,滤液于60℃下浓缩至100l得提取浓缩液(约21.35kg);
[0093]
b、树脂富集:将步骤a浓缩液加入d101树脂100kg,采用静态吸附方法吸附20h,吸附后过滤出d101树脂并装柱,依次采用水、20%乙醇、40%乙醇和95%乙醇水梯度洗脱,每个梯度洗脱3倍体积,收集40%乙醇含目标组分,远志呫吨酮ⅲ目标部分于60℃下减压浓缩至流浸膏(约8.42kg);
[0094]
c、正相硅胶富集:将步骤b的远志呫吨酮ⅲ的流浸膏用正相硅胶(青岛海洋,100-200目,5kg):硅藻土=1:1拌样,于60℃烘干24h,用粉碎机粉碎过筛,备用。烘干后上样于硅胶柱层析中(20kg,青岛海洋,100-200目),乙酸乙酯-甲醇-水=8:1:0.3-1:1:0.3梯度洗脱,tlc检测,收集远志呫吨酮ⅲ目标流分,合并浓缩,得远志呫吨酮ⅲ的正相浸膏(2.65kg);
[0095]
d、聚酰胺纯化:将步骤c所获得的远志呫吨酮ⅲ的正相浸膏拌样于聚酰胺(1kg,浙江台州四甲生化,100-200目),于60℃烘干24h后,上样于已装好的聚酰胺正相柱(5kg,浙江台州四甲生化,100-200目)层析柱中,乙酸乙酯-甲醇-水=4:1:0.2-1:1:0.2梯度洗脱,tlc检测,收集远志呫吨酮ⅲ目标流分,合并并于40℃下降压浓缩,得远志呫吨酮ⅲ的正相聚酰胺浸膏(760g);
[0096]
e、中压纯化:将步骤d所获得的远志呫吨酮ⅲ的正相聚酰胺浸膏,用约1l的70%甲醇水溶解,上样于中压反相c18柱层析柱(苏州汇通色谱公司,260
×
600mm,填料为大曹ods-rps,粒径40-60μm),依次用5%,10%,20%,50%乙腈水梯度洗脱,流速300ml/min,先用5%乙腈水洗脱2倍柱体积,除去大极性杂质,再用10%乙腈水洗脱4倍柱体积,再用20%乙腈水洗脱5倍柱体积,目标组分存在于20%乙腈水洗脱液中,最后用50%乙腈水冲出其它杂质,
hplc检测合并,收集远志呫吨酮ⅲ目标流分,合并浓缩冻干,得到hplc约90%的远志呫吨酮ⅲ的半成品(84.1g);
[0097]
f、溶剂法处理:将步骤e得到的远志呫吨酮ⅲ的半成品研磨成粉末状,依次用水、甲醇结晶,反复操做3-5次,过滤得沉淀,hplc纯度大于99%,参照图5;
[0098]
g、样品干燥:将步骤f过滤后沉淀放入干净的容器中,55℃真空干燥8h,期间不断搅拌使样品受热均匀,干燥后将样品取出研磨成均匀的粉末,55℃再次真空干燥12h,干燥后得纯品26g。
[0099]
获得的远志呫吨酮ⅲ的nmr图谱参考图3和图4,质谱图谱参见图2。
[0100]
比较例1
[0101]
取与实例1同一产地的中药材远志100kg,前处理阶段与实施例1步骤a、b、c、d、e相同,将步骤e获得的远志呫吨酮ⅲ的半成品用100ml甲醇水溶解,进行高压反相色谱制备(使用的设备为岛津lc-20ap,为岛津企业管理(中国)生产,制备柱型号ymc-triart c18,50
×
250mm,7μm),以17%乙腈水等度洗脱,检测波长320nm,收集目标组分,hplc纯度仅有95%,达不到99%,参照图6。
[0102]
上述披露的各技术特征并不限于已披露的与其它特征的组合,本领域技术人员还可根据发明之目的进行各技术特征之间的其它组合,以实现本发明之目的,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1