一种环烯基在β位取代的吖内酯衍生物的制备方法与流程
一种环烯基在
β
位取代的吖内酯衍生物的制备方法
技术领域
1.本发明属于医药中间体合成技术领域,具体涉及一种环烯基在β位取代的吖内酯衍生物的制备方法,该化合物是制备雷米普利的关键中间体。
背景技术:
2.雷米普利是一种非巯基的血管紧张素转化酶抑制剂。作为前体药物,经胃肠道吸收后在肝脏水解生成有活性的雷米普利拉。临床上用于原发性高血压、充血性心力衰竭、肾性高血压及急性心梗发作后前几天之内出现的充血性心力衰竭症状者。(s,s,s)-2-氮杂双环[3,3,0]辛烷-3-羧酸是合成雷米普利的关键中间体。
[0003]
现有技术合成方法中有化学拆分法(例如ep115345)、生物转化法(us 2009/0017509)、不对称合成法(wo 2011/133651)。其中不对称合成法具有原子经济性好、副产物少等特点。专利wo 2011/133651报道了以环戊酮为主要起始原料合成(s,s,s)-2-氮杂双环[3,3,0]辛烷-3-羧酸。但在研究中发现该工艺中由环戊酮和三氯氧磷合成如式(
ⅴ
)所示的中间体2-氯-1-环戊烯醛,反应过程产生了大量含磷废水难以处理,不符合绿色环保要求,且中间体2-氯-1-环戊烯醛易挥发、不稳定,在加热浓缩过程中有一定安全隐患;中间体2-氯-1-环戊烯醛的结构式如下所示:
[0004][0005]
上述文献wo 2011/133651中所提合成路线:
[0006][0007]
合成式(ⅰ)所示的吖内酯衍生物经典方法是在erlenmeyer-azlactone synthesis的反应条件下反应。如上所述,醛与酰基甘氨酸在乙酸酐和碱的存在下缩合反应。但该方法需要高温长时间反应,不稳定的2-氯-1-环戊烯醛(
ⅴ
)会被破环形成焦油状物,影响反应收率。
[0008][0009]
2009年,tetrahedron,2009,65,2935-2938.报道了把醛和噁唑酮吸附于十倍当量的干燥al2o3中,进行固相催化反应。该反应利用了噁唑酮的高反应性,实现了在温和的条件下合成吖内酯衍生物,但该方法在实际操作中并不适用于工业化生产。
[0010][0011]
2018年,abdulhamid fadavi提出由vilsmeier试剂参与的苯甲醛与2-苯基-5-噁唑酮合成吖内酯衍生物(comptes rendus chimie,2018,21,9-13)。
[0012]
其中vilsmeier试剂起到了脱水剂的作用,反应条件较为温和。
[0013][0014]
针对合成方法中,环境不友好、反应物不稳定、三废产生量大,以及苛刻的反应条件等问题,开发一条条件温和、工艺简单、经济环保的新合成工艺显得尤其重要。
技术实现要素:
[0015]
针对现有技术中存在的上述问题,本发明的目的在于提供一种以式(ⅱ)为起始原料制备环烯基取代的吖内酯衍生物的方法。该方法革除了三氯氧磷的使用,避免了合成不稳定的2-氯-1-环戊烯醛,具有工艺简单、操作方便、收率较高、成本较低等特点。
[0016]
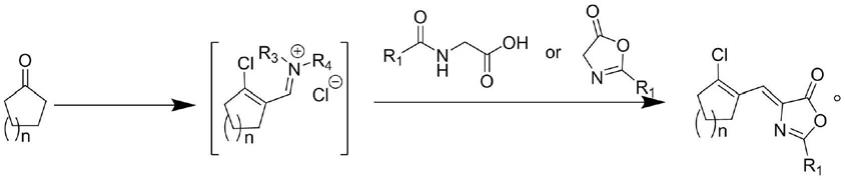
本发明限定了一种环烯基在β位取代的吖内酯衍生物的制备方法,所述环烯基取代的吖内酯衍生物的结构式如式(ⅰ)所示,其特征在于以式(ⅱ)所示的化合物和式(ⅲ)所示的氨基酸衍生物或式(ⅳ)所示的吖内酯为原料,在n,n-二取代甲酰胺衍生物和双(三氯甲基)碳酸酯存在下反应制得式(ⅰ)所示的化合物;
[0017][0018][0019]
式中:n为1,2,3或4;r1为烷基或苯基。
[0020]
进一步地,本发明限定了所述环烯基在β位取代的吖内酯衍生物的制备方法,具体包括以下步骤:
[0021]
1)将式(ⅱ)所示化合物、n,n-二取代甲酰胺衍生物溶于溶剂a中得混合溶液,向混合溶液中滴加双(三氯甲基)碳酸酯与溶剂a的溶液,至式(ⅱ)所示的化合物消失,得到铵盐过渡产物;
[0022]
2)将式(ⅲ)所示的氨基酸衍生物或式(ⅳ)所示的吖内酯加入到步骤1)的铵盐过渡产物中,控温下进行反应,tlc监测至反应物消失,反应结束后浓缩除去溶剂,用溶剂b打浆纯化得到式(ⅰ)化合物;
[0023][0024]
其中n为1,2,3或4;r1为烷基或者苯基。
[0025]
进一步地,本发明还限定了步骤1)中的溶剂a为氯仿、二氯甲烷、1,2-二氯乙烷、正己烷、环己烷、环戊烷、甲基环己烷、四氢呋喃或甲基叔丁基醚。
[0026]
进一步地,本发明还限定了步骤1)中的反应温度为-20℃~60℃。
[0027]
进一步地,本发明还限定了步骤1)中的n,n-二取代甲酰胺衍生物为n,n-二甲基甲酰胺、n-甲基甲酰苯胺或n,n-二甲基乙酰胺。
[0028]
进一步地,本发明还限定了步骤2)中的溶剂b为甲醇、乙醇、乙酸乙酯或二氯甲烷。
[0029]
进一步地,本发明还限定了步骤2)中的反应温度为0~90℃。
[0030]
进一步地,本发明还限定了式(ⅳ)所示的化合物的制备方法,它可以由式(ⅲ)所示的氨基酸衍生物通过脱水缩合反应得到,具体为:以式(ⅲ)所示的氨基酸衍生物为原料,在脱水剂存在下,于一定温度下发生脱水反应,反应结束后得到式(ⅳ)所示的吖内酯。
[0031]
进一步地,本发明还限定了脱水剂为乙酸酐、二环己基碳二亚胺、1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐或氯代亚甲基铵盐;脱水剂与式(ⅲ)所示的氨基酸衍生物的物质的量之比为1.0~3.0:1。
[0032]
进一步地,本发明还限定了脱水缩合反应的温度为15-40℃。
[0033]
本发明的反应过程如下:
[0034][0035]
其中式(ⅰ)、(ⅱ)、(ⅲ)、(
ⅴ
)及反应通式中,n为1,2,3或4;r1、r3、r4为烷基或者各种取代苯基;
[0036]
通过采用上述技术,与现有技术相比,本发明的有益效果体现在:
[0037]
1)本发明通过采用一锅法合成目标产物,其工艺简单、反应条件更温和,能耗较低,后处理简便;
[0038]
2)本发明在合成过程中避免了使用三氯氧磷,消除含磷废水的产生,环境友好,产生的三废少,产物收率和纯度较高,适于工业化生产。
具体实施方式
[0039]
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
[0040]
实施例1:制备2-苯基-5-噁唑酮(
ⅳ‑
1)
[0041]
以r1为苯基的具体实施例,按照以下合成路线进行制备2-苯基-5-噁唑酮:
[0042][0043]
在带有磁力搅拌、温度计的50ml三口烧瓶中加入n-苯甲酰甘氨酸(1.79g,10mmol)、二环己基碳二亚胺(3.09g,15mmol)和二氯甲烷20ml,于15℃下进行脱水反应,直至溶液中白色不溶固体消失,反应结束,过滤,旋干溶剂,得粗品1.52g,粗品产率94.4%。不作纯化,直接作下一步反应。1h nmr(400mhz,cdcl3):δ8.04
–
7.96(m,2h),7.59(t,j=7.2,1h),7.49(t,j=7.2,2h),4.10(s,2h)。
[0044]
实施例2:制备2-苯基-5-噁唑酮(
ⅳ‑
1)
[0045]
在带有磁力搅拌、温度计的25ml三口烧瓶中加入n-苯甲酰甘氨酸(0.89g,5.0mmol)、乙酸酐(1.53g,15.0mmol),加热至温度在70℃左右进行脱水反应,直至溶液中白色不溶固体消失,反应结束。旋蒸除去乙酸,水洗除去剩余酸酐,得粗品0.68g,收率85.5%,不作进一步纯化,直接作下一步反应。
[0046]
实施例3:制备2-苯基-5-噁唑酮(
ⅳ‑
1)
[0047]
在带有磁力搅拌、温度计的50ml三口烧瓶中加入n-苯甲酰甘氨酸(1.79g,10mmol)、1-乙基-(3-二甲基氨基丙基)碳化二亚胺盐酸盐(2.10g,11mmol)和二氯甲烷20ml,于35℃下进行脱水反应,直至溶液中白色不溶固体消失,反应结束。除去溶剂,得粗品1.55g,粗品产率96.3%。不作纯化,直接作下一步反应。
[0048]
实施例4:制备2-苯基-5-噁唑酮(
ⅳ‑
1)
[0049]
在带有磁力搅拌、温度计的50ml三口烧瓶中加入n,n-二甲基甲酰胺(0.73g,10mmol)和二氯甲烷(10ml),降温至15℃,缓慢滴加双(三氯甲基)碳酸酯(0.99g,3.3mmol)的二氯甲烷(10ml)溶液,控制反应温度不超过15℃,生成氯代亚甲基铵盐作为脱水剂,滴加结束后加入n-苯甲酰甘氨酸(1.79g,10mmol)的n,n-二甲基甲酰胺(5ml)溶液,于55℃下反应,tlc监测至反应结束,展开剂配比为正己烷:乙酸乙酯:乙酸=1:1:0.05。除去溶剂,得粗品1.02g,粗品产率63.3%。不作纯化,直接作下一步反应。
[0050]
实施例5:制备4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮(
ⅰ‑
1)
[0051]
以n=1,r1=ph为具体实施例,按照以下合成路线进行制备4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮
[0052][0053]
在带有磁力搅拌、温度计、滴液漏斗的50ml三口烧瓶中加入环戊酮(0.84g,10mmol)、双(三氯甲基)碳酸酯(1.98g,6.7mmol)和二氯甲烷20ml,降温至10℃,缓慢滴加n,n-二甲基甲酰胺(1.46g,20mmol),滴加过程使反应温度不超过30℃,滴加结束后继续保温反应,跟踪反应至原料环戊酮反应完全,加入实例1中制备的2-苯基-5-噁唑酮,tlc监测至反应结束,展开剂配比为正己烷:乙酸乙酯=2:1。浓缩除去二氯甲烷,加入乙醇打浆,过滤烘干后得到黄色固体4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮2.45g,收率90.0%,hplc纯度99.5%。1h nmr(400mhz,cdcl3):δ8.12
–
8.05(m,2h),7.62
–
7.55(m,1h),7.46-7.54(m,2h),7.28(s,1h),3.09-3.20(m,2h),2.73-2.83(m,2h),2.04-2.15(m,2h).
[0054]
实施例6:制备4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮(
ⅰ‑
1)
[0055]
以n=1,r1=ph为具体实施例:
[0056]
在带有磁力搅拌、温度计、滴液漏斗的50ml三口烧瓶中加入环戊酮(0.42g,5.0mmol)、双(三氯甲基)碳酸酯(0.89g,3.0mmol)和环己烷20.0ml,降温至0℃,缓慢滴加n,n-二甲基甲酰胺(10.0mmol,0.73g),滴加过程使内部温度不超过28℃,滴加结束保温反应一小时,升温至38℃,跟踪反应至原料环戊酮反应完全,加入实例2中制备的2-苯基-5-噁唑酮,tlc监测至反应结束,展开剂配比为正己烷:乙酸乙酯=2:1。浓缩除去二氯甲烷,加入乙酸乙酯打浆,过滤烘干后得到黄色固体4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮1.1g,收率80.5%。
[0057]
实施例7:制备4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮(
ⅰ‑
1)
[0058]
以n=1,r1=ph为具体实施例:
[0059]
在带有磁力搅拌、温度计、滴液漏斗的50ml三口烧瓶中加入环戊酮(0.84g,10mmol)、双(三氯甲基)碳酸酯(1.48g,5.0mmol)和二氯甲烷20.0ml,降温至5℃,缓慢滴加
n,n-二甲基乙酰胺(15mmol,1.30g),滴加过程使内部温度不超过25℃,滴加结束后升温至35℃继续反应,tlc跟踪反应至原料环戊酮反应完全,直接加入n-苯甲酰甘氨酸,tlc监测至反应结束,展开剂配比为正己烷:乙酸乙酯=2:1。浓缩除去二氯甲烷,加入甲醇打浆,过滤烘干后得到黄色固体4-((2-氯环戊基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮1.22g,收率34.4%。
[0060]
实施例8:制备4-((2-氯环己基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮(
ⅰ‑
2)
[0061]
以n=2,r1=ph为具体实施例:
[0062]
在带有磁力搅拌、温度计、滴液漏斗的50ml三口烧瓶中加入环己酮(0.98g,10mmol)、双(三氯甲基)碳酸酯(2.07g,7.0mmol)和二氯甲烷20.0ml,降温至0℃,缓慢滴加n-甲基甲酰苯胺(2.97g,22mmol),滴加结束后于20℃保温反应一小时,升温至30℃,跟踪反应至原料环己酮反应完全,加入实例1中制备的2-苯基-5-噁唑酮,tlc监测至反应结束,展开剂配比为正己烷:乙酸乙酯=2:1。浓缩除去二氯甲烷,加入乙醇打浆,过滤烘干后得到黄色固体4-((2-氯环己基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮2.53g,收率88.0%。1h nmr(400mhz,cdcl3)δ8.14
–
8.05(m,2h),7.61
–
7.57(m,1h),7.56(s,1h),7.48-7.51(m,2h),2.96-2.99(m,2h),2.61-2.63(m,2h),1.75-1.82(m,4h)。
[0063]
实施例9:制备4-((2-氯环己基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮(
ⅰ‑
2)
[0064]
以n=2,r1=ph为具体实施例:
[0065]
在带有磁力搅拌、温度计、滴液漏斗的50ml三口烧瓶中加入环己酮(0.98g,10mmol)、双(三氯甲基)碳酸酯(2.50g,8.4mmol)和二氯甲烷20.0ml,降温至0℃,缓慢滴加n,n-二甲基甲酰胺(1.825g,25mmol),滴加结束后升温至40℃,跟踪反应至原料环己酮反应完全,直接加入n-苯甲酰甘氨酸,tlc监测至反应结束,展开剂配比为正己烷:乙酸乙酯=2:1。浓缩除去二氯甲烷,加入甲醇打浆,过滤烘干后得到黄色固体4-((2-氯环己基-1-烯基)亚甲基)-2-苯基噁唑-5-(4h)-酮1.15g,收率40.0%。
[0066]
最后应当说明的是,以上实施仅用以说明本发明的技术方案而非限制本发明,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1