一类雷公藤甲素前药、其制备方法及其医药用途
1.本发明属于药物化学领域,涉及一类雷公藤甲素水溶性前药或其药学上可接受的盐,以及含有这些化合物的药物组合物,它们的制备方法以及医药用途。
背景技术:
2.雷公藤(tripterygium wilfordii hook.f.)为卫矛科(celastraceae)雷公藤属植物,具有祛风除湿、活血通络、消肿止痛、杀虫解毒等功效,是临床治疗自身免疫性疾病的首选中药。目前已经有多种雷公藤提取物药物如雷公藤多苷片、雷公藤片(999)、雷公藤双层片、雷公藤总萜片等上市应用于类风湿性关节炎、自身免疫性肝炎、肾炎及肾病综合征等多种免疫性和炎症性疾病的治疗。雷公藤提取物的主要活性成分有二萜类、三萜类和生物碱类,其中二萜类的雷公藤甲素是雷公藤的主要活性成分之一,同时也是雷公藤多苷片、雷公藤片等制剂的主要有效成分。
3.作为雷公藤的高活性成分之一,雷公藤甲素具有较强的免疫抑制、抗炎、抗肿瘤等多种药理作用【j am chem soc,1972,94(20):7194-7195;drugs r d,2003,4(1):1-18;trendspharmacol sci,2019,40(5):327-341】。自1969年以来,中药雷公藤一直被广泛用于治疗类风湿性关节炎,美国风湿病学会一项双盲临床研究同样显示雷公藤可以明显改善类风湿性关节炎症状,有效率达到58%【j rheumatol 2003,30(3):465-467】。对于其他自身免疫性以及炎症性疾病,如系统性红斑狼疮、银屑病、强直性脊柱炎、哮喘、肾炎、溃疡性结肠炎、肺纤维化等,雷公藤均表现出令人满意的效果【chin j mod appl pharm,1999,16(2):10-13; br j clin pharmacol 2012sep;74(3):424-36;rheum dis clin north am 2000;am j pathol 2001; 158(3):997-1004;26(1):29-50】。鉴于雷公藤的免疫抑制作用,许多学者研究发现雷公藤甲素在心脏、肾脏、肝脏或骨髓移植后,可有效阻止移植器官引起的排斥反应,显著延长动物的存活期【transplantation 2000;70(3):447-55;transplant proc 1999;31(7):2719-23】。在癌症领域,雷公藤甲素通过共价结合并抑制tfhii转录复合体中xpb活性从而调控细胞的整体转录水平,使雷公藤甲素具有广谱的抗肿瘤活性【angew chem int ed engl,2015,54(6): 1859-1863;mol cancer ther,2003,2:65
–
72】。此外,雷公藤甲素在体外表现出有效的抗hiv 作用【j nat prod 2000;63(3):357-61】,并且多项临床实现正在进行【nct03403569; nct01817283;nct02002286】。然而,由于其二萜内酯类的结构水溶性较差,限制了其临床应用。因此,可以通过引入水溶性基团,设计合成雷公藤甲素的水溶性前药,在不影响其药效的情况下,提高其水溶性、改善其成药性。
4.水溶性前药的设计策略主要是在雷公藤甲素的c-14位通过酯键或缩醛连接一些水溶性基团。如pg490-88是在雷公藤甲素的c-14位直接通过酯键引入水溶性的羧酸,是第一个进入临床研究的雷公藤甲素前药【us5663335】。临床试验中虽然大多数患者的毒副作用是可以控制的,但有两个患者出现了致命副作用,其中一名在12mg/kg剂量下死于中性粒细胞减少性败血症,另一名在18mg/kg剂量下死于复杂的临床综合征。后续的药代动力学实验发现, pg490-88在体内无法迅速转化为雷公藤甲素,其不仅在不同物种(包括小鼠、猴子
和人) 中出现了很大的差异性,在同一批18名志愿者中也表现出了2-3倍的转化差异。究其原因,可能是pg490-88的c-14位空间位阻较大,阻碍了酯酶对酯键的水解,导致pg490-88在体内转化缓慢且不完全。此外,由于个体之间酯酶活性存在差异,也导致药物的安全剂量难以控制。因此,pg490-88的i期临床试验也被迫于2009年终止。
5.为了克服pg490-88体内转化不完全的问题,科研工作者们致力于对pg490-88的c-14位的连接方式进行优化,以避免空间位阻对断键的影响。2010年,georg等人以甲缩醛作为连接臂引入磷酸基团设计合成了水溶性雷公藤甲素前药minnelide【wo2010/129918】。由于磷酸酯键旁的空间位阻较小,minnelide很容易被磷酸酯酶水解生成羟甲基中间体,中间体羟甲醚的结构很不稳定,会自动水解释放出c14位的羟基。因此,minnelide在i期临床试验中表现出较好的药代动力学性质,已经进入了ii期临床试验。
[0006][0007]
虽然甲缩醛的结构容易水解断裂,但在体外人血浆转化实验中我们发现minnelide完全转化成雷公藤甲素仍然需要24小时以上,可能与血浆中碱性磷酸酯酶含量不高有关。此外,由于雷公藤甲素原料价格昂贵,minnelide的合成难度较大、总收率较低(39%),使其开发成本较高;且由于反应条件较苛刻,不利于规模化制备。因此,开发一种水溶性好、能够快速完全转化、并且合成工艺简单的雷公藤甲素前药具有重要的市场前景。
[0008]
羧酸酯酶是人和动物体内含量最丰富的水解酶,参与多种内源性和外源性化合物及药物的代谢过程,本发明通过羟基酸酯(一种容易被酯酶水解的结构)的连接方式在雷公藤甲素的c-14位引入水溶性的脂肪性含氮杂环,发现了一类水溶性好、体内转化迅速、并且易于合成(总收率达到61%)的雷公藤甲素水溶性前药。该前药水溶性好,口服有效,在人血浆中可以在1小时内完全转化成雷公藤甲素,因此,其适用于雷公藤甲素有效治疗的各种炎症性疾病、免疫性疾病、血液系统恶性肿瘤和实体瘤的治疗。
技术实现要素:
[0009]
发明目的:本发明的目的在于提供了一种通式(i)所示的水溶性显著提高并且在血浆中可以迅速转化的雷公藤甲素前药或其药学上可接受的盐。
[0010]
技术方案:本发明所述的一类通过易于自降解的羟基酸酯连接臂引入脂肪性含氮杂环的雷公藤甲素前药或其药学上可接受的盐、多晶型物或溶剂化物;所述衍生物的化学结构式如式(i)所示:
[0011][0012]
其中:
[0013]
n1是1~6;
[0014]
n2是1~6;
[0015]
n3是0或1;
[0016]
x为碳、氮或氧原子;
[0017]
ha选自盐酸、硫酸、碳酸、柠檬酸、琥珀酸、酒石酸、磷酸、乳酸、丙酮酸、乙酸、马来酸、甲磺酸、苯磺酸、对甲苯磺酸或阿魏酸;
[0018]
本发明优选的典型化合物如下(所述的雷公藤甲素前药选自下列化合物),但不限于:
[0019][0020][0021]
本发明的另一目的在于提供一种制备该类雷公藤甲素前药或其药学上可接受的
盐、多晶型物或溶剂化物的制备方法;其具体制备步骤如下:
[0022]
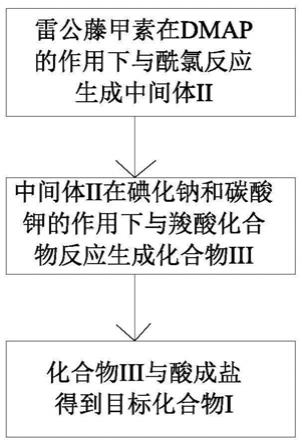
第一步:雷公藤甲素在dmap的作用下与酰氯反应生成中间体ii;
[0023]
将雷公藤甲素和dmap溶于无水二氯甲烷中,在-10~5℃的低温条件下加入酰氯,后将其置于0~30℃的温度下反应6~12小时;再将得到的反应液依次用稀盐酸、饱和碳酸氢钠和盐水洗,无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化得到中间体ii;
[0024][0025]
第二步:中间体ii在碘化钠和碳酸钾的作用下与相应的羧酸化合物反应生成化合物iii;
[0026]
将纯化得到的中间体ii溶于无水dmf中,后加入碘化钠和羧酸,进行反应0.5~1小时后加入碳酸钾,再将其加热至40~70℃反应3~12小时得反应液;然后将反应液倒入水中,用乙酸乙酯萃取,碳酸氢钠水溶液和食盐水洗涤,干燥,抽滤,浓缩滤液,柱层析纯化得到化合物iii;
[0027][0028]
第三步:化合物iii与相应的酸成盐得到目标化合物i;
[0029]
将纯化得到的化合物iii溶于乙酸乙酯中,加入无机酸或有机酸,于0~30℃的温度下反应6~12小时,抽滤,干燥得到目标化合物i。
[0030][0031]
本发明的目的在于提供一种药物组合物,包括所述的雷公藤甲素前药、其药学上可接受的盐、多晶型物或溶剂化物,以及至少一种药学上可接受的载体、添加剂、助剂或赋形剂。
[0032]
本发明的另一目的在于提供所述雷公藤甲素前药或药学上可接受的盐、及其药物组合物在制备抗肿瘤药物中的用途。
[0033]
本发明所述的雷公藤甲素前药在人血浆中可以完全转化成雷公藤甲素,因此,本发明提供的雷公藤甲素前药或其药学上可接受的盐,及其药物组合物可以可作为单一治疗剂,或者与其它抗肿瘤药物联合使用,用于雷公藤甲素有效的多种恶性肿瘤的治疗,具体包括急性髓细胞白血病、淋巴瘤、骨髓瘤、肺癌、肝癌、乳腺癌、结直肠癌、卵巢癌、宫颈癌、胰腺癌、胆管癌、胃癌、前列腺癌、肾癌、食管癌、胶质母细胞瘤以及成神经细胞瘤等恶性肿瘤等多种恶性肿瘤。
[0034]
本发明的另一目的在于提供所述雷公藤甲素前药或药学上可接受的盐、及其药物
组合物在制备治疗急性髓细胞白血病药物中的用途;体外抗肿瘤活性实验表明,本发明的化合物能够显著抑制急性髓细胞白血病细胞的增殖;整体动物试验表明,本发明化合物对急性髓细胞白血病具有很好的疗效,起效剂量仅为25μg/kg,并且与flt3抑制剂联合使用具有协同增效作用;因此,本发明的化合物或其药学上可接受的盐,及其药物组合物可作为单一治疗剂,或者与flt3抑制剂联合使用,用于急性髓细胞白血病的治疗。
[0035]
本发明的另一目的在于提供所述雷公藤甲素前药或药学上可接受的盐、及其药物组合物在制备抗炎、免疫抑制及病毒(例如:抗hiv)药物中的用途;本发明所述的雷公藤甲素前药在人血浆中可以完全转化成雷公藤甲素;因此,本发明提供的雷公藤甲素前药或其药学上可接受的盐,及其药物组合物可以用于雷公藤甲素有效的各种适应症的治疗,包括类风湿性关节炎、强直性脊柱炎、系统性红斑狼疮、系统性血管炎、银屑病、特发性皮炎、炎症性肠病、哮喘、肺纤维化、肾炎、肾病综合征、免疫排斥反应、以及lps诱导、car-t疗法、细菌感染、病毒感染等引起的细胞因子释放综合征。
[0036]
有益效果:本发明与现有技术相比,本发明的特点是:本发明所述的雷公藤甲素前药具有显著的体内抗肿瘤和免疫抑制活性,相比雷公藤甲素具有更好的水溶性和药代动力学性质,相比minnelide,不仅体内转化更迅速,而且合成更简单,收率较高(61%),成本更低,具有良好的产业化前景。
附图说明
[0037]
图1是本发明的制备流程图;
[0038]
图2是本发明实施例11中tp-p1、tp-p2和tp-p5在大鼠血浆中的体外转化实验的示意图;
[0039]
图3是本发明实施例12中tp-p1、tp-p5在人血浆中的体外转化实验的示意图;
[0040]
图4是本发明实施例13中tp-p1、pg490-88na、minnelide在大鼠血浆中的体外转化实验的示意图;
[0041]
图5是本发明实施例14中tp-p1、pg490-88na、minnelide在人血浆中的体外转化实验的示意图;
[0042]
图6是本发明实施例15中不同浓度tp-p1在人血浆中的体外转化实验的示意图。
具体实施方式
[0043]
为了更清楚地说明本发明的技术方案,下面结合附图对本发明的技术方案做进一步的详细说明:
[0044]
实施例1
[0045]
化合物tp-p1的合成
[0046][0047]
中间体ii-1的合成:将雷公藤甲素tp(1.8g,5.0mmol)溶于50ml无水二氯甲烷中,在 0℃下加入dmap(3.05g,25.0mmol),然后逐滴加入氯乙酰氯(4.0ml,50.00mmol),滴加完
毕后,于25℃反应12小时;反应完毕后,将反应液依次用5%的稀盐酸、饱和碳酸氢钠和饱和氯化钠溶液洗涤,然后使用无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=200:1~100:1)得到白色固体1.77g,收率88.8%。1h nmr(500mhz,meod)δ(ppm): 5.14(1h,s),4.78-4.85(2h,m),4.31(2h,d,j=1.0hz),3.99(1h,d,j=3.2hz),3.67(1h,d,j= 2.6hz),3.53(1h,d,j=5.7hz),2.79-2.81(1h,m),2.24-2.32(2h,m),2.07-2.12(1h,m), 1.88-1.98(2h,m),1.50-1.54(1h,m),1.31-1.39(1h,m),1.05(3h,s),0.98(3h,d,j=7.0hz), 0.86(3h,d,j=6.9hz).
[0048][0049]
中间体iii-1的合成:将化合物ii-1(1.0g,2.3mmol)溶于50ml无水dmf中,加入碘化钠(860mg,4.6mmol)和吗啉-4-基乙酸(670mg,4.6mmol);室温反应40分钟后,加入碳酸钾(320mg,2.3mmol),然后加热至50℃反应4小时;反应完毕后,将反应液倒入水中,乙酸乙酯萃取,合并有机层,依次用碳酸氢钠水溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=100:1~50:1)得到白色固体1.01g,收率 80.4%;1h nmr(500mhz,meod)δ(ppm):5.13(1h,s),4.82-4.85(2h,m),4.77-4.79(2h,m), 3.99(1h,d,j=3.2hz),3.72-3.75(4h,m),3.65(1h,d,j=2.5hz),3.51(1h,d,j=2.5hz), 3.43(2h,s),2.79-2.81(1h,m),2.63-2.67(4h,m),2.23-2.32(2h,m),2.04-2.10(1h,m), 1.88-1.98(2h,m),1.49-1.54(1h,m),1.31-1.39(1h,m),1.05(3h,s),0.98(3h,d,j=10.7hz), 0.86(3h,d,j=6.9hz).
[0050][0051]
合物tp-p1的合成:将化合物iii-1(1.0g,1.83mmol)溶于20ml乙酸乙酯中,加入20 ml饱和的氯化氢的乙酸乙酯溶液,于室温反应6小时;反应完成后,抽滤,滤饼用乙酸乙酯洗涤,然后真空干燥得到白色固体0.91g,收率85.3%。1h nmr(500mhz,dmso-d6)δ (ppm):10.93(1h,s),5.46(1h,s),4.88-4.92(2h,m),4.77-4.84(2h,m),4.59(1h,m),4.37(2h, s),3.88(1h,d,j=4.9hz),3.72-3.85(4h,m),3.56(1h,d,j=5.5hz),3.05-3.30(4h,m), 2.64-2.74(1h,m),2.15-2.24(2h,m),1.95-1.98(2h,m),1.80-1.88(1h,m),1.36-1.41(1h,m), 1.25-1.32(1h,m),0.93(3h,d,j=6.5hz),0.90(3h,s),0.78(3h,d,j=6.6hz).
13
c nmr(126 mhz,dmso-d6)δ(ppm):173.6,166.6,166.5,162.5,123.7,75.5,75.4(2c),70.8,66.8,62.5, 61.8,59.5(2c),58.1,57.4,52.3(2c),35.4,34.6,30.4,29.2,22.5,17.1,16.3,15.9(2c),14.6. lcms(esi):m/z[m+h]
+
calcd for c
28h37
clno
10+
,582.2;found,582.0.
[0052]
实施例2
[0053]
化合物tp-p2的合成
[0054][0055]
中间体iii-2的合成:将化合物ii-1(100mg,0.23mmol)溶于10ml无水dmf中,加入碘化钠(86mg,0.46mmol)和4-甲基-1-哌嗪乙酸(73mg,0.46mmol);于25℃反应40分钟后,加入碳酸钾(32mg,0.23mmol),然后加热至50℃反应4小时;反应完毕后,将反应液倒入水中,乙酸乙酯萃取,合并有机层,依次用碳酸氢钠水溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=100:1~40:1)得到白色固体 93mg,收率72.3%;1h nmr(500mhz,meod-d6)δ(ppm):5.12(1h,s),4.81-4.85(2h,m), 4.77-4.80(2h,m),3.98(1h,d,j=3.1hz),3.65(1h,d,j=2.8hz),3.50(1h,d,j=5.7hz), 3.47(2h,s),2.79-2.83(1h,m),2.66-2.78(8h,m),2.40(3h,s),2.26-2.32(2h,m),2.04-2.10(1h, m),1.89-1.95(2h,m),1.49-1.54(1h,m),1.31-1.39(1h,m),1.05(3h,s),0.97(3h,d,j=7.0 hz),0.86(3h,d,j=6.9hz).
[0056][0057]
化合物tp-p2的合成:将化合物iii-2(100mg,0.18mmol)溶于5ml乙酸乙酯中,加入 3ml饱和的氯化氢的乙酸乙酯溶液,室温反应6小时,反应完成后,抽滤,滤饼用乙酸乙酯洗涤,然后真空干燥得到白色固体75mg,收率70.4%;1h nmr(500mhz,meod)δ(ppm): 4.83-4.86(2h,m),4.82(2h,s),4.74(1h,s),4.32(1h,d,j=5.3hz),3.95(1h,d,j=5.4hz), 3.84-3.93(2h,m),3.59-3.71(2h,m),3.53(1h,d,j=6.1hz),3.37-3.47(2h,m),3.12-3.28(4h, m),2.96(3h,s),2.82-2.85(1h,m),2.27-2.33(2h,m),2.08-2.14(2h,m),1.93-1.99(1h,m), 1.56-1.59(1h,m),1.35-1.41(1h,m),1.05(3h,s),1.01(3h,d,j=6.8hz),0.88(3h,d,j=6.9 hz).
13
c nmr(126mhz,meod)δ(ppm):174.6,168.4,166.9,162.3,124.3,75.4,75.2,70.7, 67.0,62.2,60.5,59.3,58.0,56.8,56.0,52.6(2c),48.9(2c),42.1,39.6,35.3,30.3,28.9,22.4, 16.5,14.8,14.4,13.2.lcms(esi):m/z[m+h]
+
calcd for c
29h40
cln2o
9+
,595.2;found,595.2.
[0058]
实施例3
[0059]
化合物tp-p3的合成
[0060][0061]
化合物tp-p3的合成:将化合物ii-1(100mg,0.23mmol)溶于10ml无水dmf中,加入碘化钠(86mg,0.46mmol)和2-哌啶基乙酸(66mg,0.46mmol);于25℃反应40分钟后,加入碳酸钾(32mg,0.23mmol),然后加热至50℃反应4小时;反应完毕后,将反应液倒入水中,乙酸乙酯萃取,合并有机层,依次用碳酸氢钠水溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥;
抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=100:1~40:1)得到白色固体67mg,收率53.4%;1h nmr(500mhz,cdcl3)δ(ppm):5.11(1h,s),4.81-4.85(2h,m),4.65-4.73(2h, m),3.84(1h,d,j=3.1hz),3.64(2h,d,j=4.2hz),3.56(1h,d,j=2.8hz),3.48(1h,d,j= 5.7hz),2.87-2.98(4h,m),2.32-2.36(1h,m),2.16-2.26(2h,m),1.89-1.95(2h,m),1.76-1.80 (4h,m),1.52-1.59(2h,m),1.32-1.38(2h,m),1.06(3h,s),0.97(3h,d,j=7.0hz),0.86(3h,d, j=6.9hz).
13
c nmr(126mhz,cdcl3)δ(ppm):173.2,168.0,167.0,159.9,125.7,72.4,69.9, 63.5,63.1,61.3,60.8,59.4,57.9,55.4,55.0,53.4(2c),40.3,35.7,29.9,28.0,24.6(2c),23.4, 23.0,17.5,17.1,16.7,13.8.lcms(esi):m/z[m+h]
+
calcd for c
29h38
no
9+
,544.2;found,544.3.
[0062]
实施例4
[0063]
化合物tp-p4的合成
[0064][0065]
化合物tp-p4的合成:将化合物ii-1(100mg,0.23mmol)溶于无水10ml dmf中,加入碘化钠(86mg,0.46mmol)和2-(吡咯烷-1-基)乙酸(59mg,0.46mmol);于25℃反应40分钟后,加入碳酸钾(32mg,0.23mmol),然后加热至50℃反应4小时;反应完毕后,将反应液加入水中,乙酸乙酯萃取,合并有机层,依次用碳酸氢钠水溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=100:1~40:1)得到白色固体 64mg,收率52.3%;1h nmr(500mhz,meod)δ(ppm):5.14(1h,m),4.81-4.85(2h,m), 3.98-4.03(2h,m),3.73(1h,m),3.66(1h,d,j=7.3hz),3.51(1h,d,j=5.7hz),3.27(2h,s), 3.14-3.18(1h,m),2.79-2.81(1h,m),2.59-2.62(1h,m)2.22-2.32(2h,m),2.02-2.09(4h,m), 1.85-1.92(2h,m),1.49-1.53(1h,m),1.35-1.42(4h,m),1.05(3h,s),0.97(3h,d,j=7.0hz), 0.86(3h,d,j=6.9hz).
13
c nmr(126mhz,meod)δ(ppm):174.6,172.4,162.4,161.9,124.1, 71.7,70.6,65.9,63.5,62.7,61.4,59.6,59.5,55.4,54.8,54.3,52.9,40.0,35.4,29.4,28.2,22.8 (2c),22.7,16.5(2c),15.7,12.7.lcms(esi):m/z[m+h]
+
calcd for c
28h36
no
9+
,530.2;found, 530.2.
[0066]
实施例5
[0067]
化合物tp-p5的合成
[0068][0069]
中间体ii-5的合成:将雷公藤甲素(180mg,0.50mmol)溶于20ml无水二氯甲烷中,在0℃条件下加入dmap(305mg,2.50mmol),然后逐滴加入4-溴丁酰氯(0.56ml,5.00 mmol),滴加完毕后,于25℃反应12小时;反应完毕后,将反应液依次用5%的稀盐酸、饱和碳酸氢钠和饱和氯化钠溶液洗涤,然后使用无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=200:1~100:1)得到白色固体193mg,收率75.6%。1h nmr(500mhz, cdcl3)δ
(ppm):5.11(1h,s),4.65-4.73(2h,m),4.33(1h,t,j=7.1hz),3.85(1h,d,j=2.9hz), 3.52-3.55(2h,m),3.49(1h,d,j=5.6hz),2.60-2.72(4h,m),2.51(1h,t,j=8.1hz),2.24-2.30 (3h,m),1.89-1.98(2h,m),1.57-1.62(1h,m),1.26-1.35(1h,m),1.07(3h,s),0.98(3h,d,j= 6.9hz),0.87(3h,d,j=6.9hz).
[0070][0071]
中间体iii-5的合成:将化合物ii-5(117mg,0.23mmol)溶于10ml无水dmf中,加入碘化钠(86mg,0.46mmol)和吗啉-4-基乙酸(67mg,0.46mmol);于25℃反应40分钟后,加入碳酸钾(32mg,0.23mmol),然后加热至50℃反应4小时;反应完毕后,将反应液倒入水中,乙酸乙酯萃取,合并有机层,依次用碳酸氢钠水溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=100:1~50:1)得到白色固体83mg,收率63.2%;1h nmr(500mhz,meod)δ(ppm):5.10(1h,s),4.78-4.84(2h,m),4.24(2h,t,j =6.4hz),3.98(1h,d,j=3.2hz),3.73(4h,t,j=4.7hz),3.65(1h,d,j=2.8hz),3.50(1h,d, j=5.7hz),3.29(2h,s),2.78-2.81(1h,m),2.61(4h,t,j=4.6hz),2.46-2.57(2h,m),2.24-2.32 (2h,m),2.09-2.12(1h,m),2.01-2.06(2h,m),1.86-1.95(2h,m),1.49-1.54(1h,m),1.31-1.39 (1h,m),1.05(3h,s),0.97(3h,d,j=7.0hz),0.86(3h,d,j=6.9hz).
[0072][0073]
化合物tp-p5的合成:将化合物iii-5(100mg,0.17mmol)溶解于5ml乙酸乙酯中,加入3ml饱和的氯化氢的乙酸乙酯溶液,于25℃反应6小时;反应完成后,抽滤,滤饼用乙酸乙酯洗涤,然后真空干燥得到白色固体53mg,收率51.3%。mp:239-241℃.1h nmr (500mhz,meod)δ(ppm):4.81-4.85(2h,m),4.73(1h,s),4.37-4.39(2h,m),4.33(1h,m), 4.14(2h,s),3.89-4.03(4h,m),3.55(1h,d,j=4.9hz),3.34(1h,d,j=5.5hz),2.83-2.86(1h, m),2.48-2.58(2h,m),2.28-2.32(2h,m),2.06-2.11(4h,m),1.94-2.04(2h,m),1.53-1.62(1h, m),1.32-1.40(3h,m),1.25-1.28(1h,m),1.05(3h,s),1.01(3h,d,j=6.0hz),0.90(3h,d,j= 6.0hz).
13
c nmr(126mhz,meod)δ(ppm):174.7,171.9,165.9,162.4,124.2,75.4,74.1,70.7, 66.9,65.1,63.7(2c),62.3,59.7,57.9,56.9,55.9,52.6(2c),39.6,35.3,30.3,30.2,29.1,23.5, 22.4,16.6,14.8,14.4,13.1.lcms(esi):m/z[m+h]+calcd for c
30h41
clno
10+
,610.2;found, 610.2.
[0074]
实施例6
[0075]
化合物tp-p6的合成
[0076]
[0077]
中间体ii-6的合成:将雷公藤甲素(180mg,0.50mmol)溶于20ml无水二氯甲烷中,在0℃下加入dmap(305mg,2.50mmol),然后逐滴加入5-溴戊酰氯(0.70ml,5.00mmol),滴加完毕后,于25℃反应12小时;反应完毕后,将反应液依次用5%的稀盐酸、饱和碳酸氢钠和饱和氯化钠溶液洗涤,然后使用无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=200:1~100:1)得到白色固体205mg,收率78.4%。1h nmr(500mhz,cdcl3)δ (ppm):5.10(1h,s),4.65-4.73(2h,m),3.84(1h,d,j=3.2hz),3.55(1h,d,j=3.0hz), 3.47-3.49(1h,m),3.43(2h,t,j=6.6hz),2.69-2.72(1h,m),2.50-2.58(1h,m),2.42(2h,t,j= 7,4hz),2.31-2.36(1h,m),2.11-2.21(2h,m),1.98-2.01(1h,m),1.90-1.96(2h,m),1.78-1.84 (2h,m),1.57-1.60(1h,m),1.22-1.29(1h,m),1.07(3h,s),0.97(3h,d,j=7.0hz),0.86(3h,d, j=6.9hz).
[0078][0079]
化合物tp-p6的合成:将化合物ii-6(120mg,0.23mmol)溶解于10ml无水dmf中,加入碘化钠(86mg,0.46mmol)和吗啉-4-基乙酸(67mg,0.46mmol);于25℃反应40分钟后,加入碳酸钾(32mg,0.23mmol),然后加热至50℃反应4小时;反应完毕后,将反应液倒入水中,乙酸乙酯萃取,合并有机层,依次用碳酸氢钠水溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=100:1~50:1)得到白色固体 88mg,收率65.1%;1h nmr(500mhz,meod)δ(ppm):5.10(1h,s),4.78-4.84(2h,m),4.19 (2h,t,j=5.5hz),3.98(1h,d,j=3.0hz),3.73(4h,t,j=4.5hz),3.65(1h,d,j=3.0hz), 3.50(1h,d,j=5.6hz),3.29(2h,s),2.78-2.81(1h,m),2.61(4h,t,j=4.6hz),2.48-2.54(1h, m),2.39-2.45(1h,m),2.26-2.31(2h,m),2.05-2.11(1h,m),1.85-1.99(2h,m),1.76-1.79(2h, m),1.50-1.54(1h,m),1.31-1.39(3h,m),1.05(3h,s),0.96(3h,d,j=7.0hz),0.86(3h,d,j= 6.9hz).
13
c nmr(126mhz,meod)δ(ppm):174.6,172.7,170.2,162.4,124.1,71.3,70.6,66.2 (2c),64.0,63.5,62.8,61.3,59.7,58.6,55.3,54.8,52.9(2c),40.1,35.4,33.2,29.4,28.3,27.5, 22.8,21.3,16.6,16.5,15.7,12.8.lcms(esi):m/z[m+h]+calcd for c
31h42
no
10+
,588.3;found, 588.3.
[0080]
实施例7
[0081]
化合物tp-p7的合成
[0082][0083]
中间体ii-7的合成:将雷公藤甲素(180mg,0.50mmol)溶于20ml无水二氯甲烷中,在0℃下加入dmap(305mg,2.50mmol),然后逐滴加入6-溴己酰氯(0.77ml,5.00mmol),滴加完毕后,于25℃反应12小时;反应完毕后,将反应液依次用5%的稀盐酸、饱和碳酸氢钠和饱和氯化钠溶液洗涤,然后使用无水硫酸钠干燥;抽滤,浓缩滤液,柱层析纯化(二氯甲烷:甲醇=200:1~100:1)得到白色固体191mg,收率71.2%。1h nmr(500mhz,cdcl3)δ (ppm):5.07
(ppm):170.5,167.2,165.0,159.8,125.7,72.3,70.0,65.1(2c),63.6,63.1,61.3,60.9,59.4,55.4, 55.0,52.6,52.3(2c),40.3,35.7,29.9,29.5,28.1,23.4,17.5,17.1,16.7,13.8.lcms(esi):m/z [m+h]+calcd for c
29h38
no
10+
,560.2;found,560.3.
[0090]
实施例9
[0091]
化合物的长期稳定性实验
[0092]
实验方法:将化合物于室温敞口放置90天以上,通过hplc归一化法测定化合物的纯度(表1)。
[0093]
表1实施例化合物的长期稳定性实验结果
[0094][0095]
结果显示:该系列化合物的化学稳定性很好,常温敞口放置90天后,经hplc-uv和1h-nmr谱分析,化合物的纯度未见明显降低。
[0096]
实施例10
[0097]
化合物tp-p1在纯水及酸性水溶液中的稳定性实验
[0098]
实验方法:将化合物tp-p1溶于ph=7的纯水及ph=4、ph=2的酸性水溶液中,采集不同时间点的样品,通过hplc归一化法测定化合物的纯度(表2)。
[0099]
表2化合物tp-p1在纯水及酸性水溶液中的稳定性实验结果
[0100][0101]
结果显示:化合物tp-p1在ph7和ph2、ph4的水溶液中6小时内纯度没有明显变化。
[0102]
实施例11
[0103]
tp-p1、tp-p2、tp-p5在大鼠血浆中的体外转化实验
[0104]
考虑到化合物的水溶性,我们选择水溶性较好的成盐化合物tp-p1、tp-p2、tp-p5进行大鼠的体外血浆转化实验。
[0105]
实验方法:取250μl大鼠空白血浆,加入等体积100μg/ml tp-p1、tp-p2、tp-p5水溶液,于恒温振荡器中60r/min、37℃孵育,于1、5、10、15、30、45、60、90min、2、4、6、8、10、12、24h取20μl含药血浆于预冷的60μl甲醇中,涡旋3min,4℃、14000rpm/min 离心10min,取上清进行hplc分析;
[0106]
液相分析方法:acn:0.1%tfa-h2o=35:65等度洗脱,流速:0.6ml/min
[0107]
液相分析方法:acn:0.1%tfa-h2o=35:65等度洗脱,流速:0.6ml/min
[0108]
实验结果如图2所述;
[0109]
结果显示:tp-p1、tp-p2和tp-p5三个前药在大鼠血浆中30min内都能完全转化生成 tp,但在相同的时间下,15min时tp-p1已经转化52.3%,而tp-p2和tp-p5不到50%,因此
tp-p1前药转化成tp的效率要稍高于tp-p2和tp-p5。
[0110]
实施例12
[0111]
tp-p1、tp-p5在人血浆中的体外转化实验
[0112]
鉴于化合物tp-p1和tp-p2都是采用乙醇酸的连接键,而相同时间内tp-p1转化速率优于tp-p5,我们选择tp-p1、tp-p5进行人的体外血浆转化实验。
[0113]
实验方法:取250μl人空白血浆,加入等体积tp-p1或tp-p5水溶液(100μg/ml或2 mg/ml),于恒温振荡器中60r/min、37℃孵育,于5、10、15、30、45、60、90min、2、 4、6、8、10、24h取20μl含药血浆于预冷的60μl甲醇中,涡旋3min,4℃、14000rpm/min 离心10min,取上清进行hplc分析。
[0114]
液相分析方法:acn:0.1%tfa-h2o=35:65等度洗脱,流速:0.6ml/min
[0115]
实验结果如图3所述;
[0116]
结果显示:tp-p1在低浓度下完全转化成tp需要45min,而高浓度下60min才完全转化成tp;tp-p5在低浓度下完全转化成tp需要45min,高浓度下90min才完全转化成tp;说明药物浓度对血浆转化存在影响,低浓度下转化速率较快。tp-p5与tp-p1相比,低浓度下完全转化成tp均需要45min,但在30min tp-p1已经转化90%,而tp-p5才不到80%,因此,同等浓度下,在人的血浆中tp-p1转化生成tp的速率快于tp-p5。
[0117]
实施例13
[0118]
tp-p1、pg490-88na、minnelide在大鼠血浆中的体外转化实验
[0119]
实验方法:取400μl大鼠空白血浆,加入等体积1μg/ml tp-p1、pg490-88na、minnelide 水溶液(血药浓度:500ng/ml),于恒温振荡器中60r/min、37℃孵育,于1、5、10、15、30、 45、60、90min、2、4、6、8、10、12、24h取40μl含药血浆于预冷的120μl甲醇(is= 1ng/ml)中,涡旋3min,4℃、14000rpm/min离心10min,取上清进行uplc-ms/ms分析。
[0120]
液相分析方法:流动相0.1%fa-h2o(a)和acn(b);流速:0.3ml/min;梯度洗脱程度:0~2min,15%b~80%b;2~3min,80%b~80%b;3~4min,80%b~15%b;4~5min,15% b~15%b;进样量:5μl;
[0121]
实验结果如图4所述;
[0122]
结果显示:tp-p1在大鼠的血浆中能够较快的转化生成tp,30min内就可以实现tp的完全转化;pg490-88na在大鼠的血浆中,90min内也可以完全转化成tp;而minnelide的转化也相对比较慢,需要6h才能实现tp的完全转化,可能是由于大鼠血浆中的磷酸酯酶相对含量较低;因此,在大鼠血浆的条件下,前药tp-p1转化成tp的速率远高于pg490-88na 和minnelide。
[0123]
实施例14
[0124]
tp-p1、pg490-88na、minnelide在人血浆中的体外转化实验
[0125]
实验方法:取400μl人空白血浆,加入等体积1μg/ml tp-p1、pg490-88na、minnelide 水溶液(血药浓度:500ng/ml),于恒温振荡器中60r/min、37℃孵育,于1、5、10、15、30、45、60、90min、2、4、6、8、10、12、24h取40μl含药血浆于预冷的120μl甲醇(is=1 ng/ml)中,涡旋3min,4℃、14000rpm/min离心10min,取上清进行uplc-ms/ms分析。
[0126]
液相分析方法:流动相0.1%fa-h2o(a)和acn(b);流速:0.3ml/min;梯度洗脱程度:0~2min,15%b~80%b;2~3min,80%b~80%b;3~4min,80%b~15%b;4~5min,
15% b~15%b;进样量:5μl;
[0127]
实验结果如图5所述;
[0128]
结果显示:tp-p1在人的血浆中也能够较快的转化生成tp,基本1h内可以完全转化;而pg490-88na在人的血浆中则转化特别缓慢,24h转化也不到15%,远远低于大鼠血浆中的转化速度;minnelide的转化也相对比较慢,24h转化接近80%,可能也是由于人血浆中的磷酸酯酶相对含量较低。因此,在人血浆的条件下,前药tp-p1转化成tp的速率远高于 pg490-88na和minnelide。
[0129]
实施例15
[0130]
不同浓度tp-p1在人血浆中的体外转化实验
[0131]
实验方法:取400μl人空白血浆,加入等体积tp-p1水溶液(10μg/ml、1μg/ml、 100ng/ml),于恒温振荡器中60r/min、37℃孵育,于1、5、10、15、30、45、60、90min、 2、4、6、8、10、12、24h取40μl含药血浆于预冷的120μl甲醇(is=1ng/ml)中,涡旋 3min,4℃、14000rpm/min离心10min,取上清进行uplc-ms/ms分析;
[0132]
液相分析方法:流动相0.1%fa-h2o(a)和acn(b);流速:0.3ml/min;梯度洗脱程度:0~2min,15%b~80%b;2~3min,80%b~80%b;3~4min,80%b~15%b;4~5min,15% b~15%b;进样量:5μl;
[0133]
实验结果如图6所述;
[0134]
结果显示:tp-p1在人的血浆中不同低浓度(50ng/ml、500ng/ml、5000ng/ml)下都能够较快的转化生成tp,1h内基本可以实现完全转化。
[0135]
实施例16
[0136]
tp-p1和tp对多种肿瘤细胞株的增殖抑制活性
[0137]
表3 tp-p1对多种肿瘤细胞的体外增殖抑制活性
[0138][0139]
实验方法:mv-4-11、thp-1、kg-1和hl-60是人源急性髓细胞白血病细胞,panc-1 为人源胰腺癌细胞,u937是人组织细胞淋巴瘤细胞,cag、arp-1、h929是人源骨髓瘤细胞,hepg2和hep3b为人源肝癌细胞,ht-29和hct-116为人源结肠癌细胞,mda-mb-231 是人源乳腺癌细胞,hela是人源宫颈癌细胞,a549是人源肺癌细胞;悬浮生长细胞采用 cck-8方法测定化合物对肿瘤细胞的体外抗增殖活性:用培养基3倍梯度稀释受试化合物至终浓度的两倍,取200μl至2ml ep管中备用;取适量处于对数生长期的细胞重悬于培养基中,等体积加入到含有受试化合物的培养基中,上下颠倒10次混匀,依次加入96孔板中,每孔100μl;于37℃、5%co2孵箱培养48h后,每孔加入10μl cck-8,继续孵育2 h;酶标仪读取od450吸光度值,重复两次实验;采用graphpad prism 8软件分析处理数据,求得ic
50
;贴壁生长细胞采用mtt方法测定化合物对实体瘤细胞的体外抗增殖活性:胰酶消化处于对数生长期的细胞,计数,取适量细胞重悬于培养液中,每孔100μl加于96孔板中,过夜培养后,每孔加入100μl 3倍梯度稀释的受试化合物或对照的培养基,于37℃、 5%co2孵箱培养48h后,每孔加入20μl mtt,继续37℃孵育4h,酶标仪读取od490 吸光度值,重复两次实验;采用graphpad prism 8软件分析处理数据,求得ic
50
值。
[0140]
结果显示:tp-p1及tp对大多数肿瘤细胞具有较强的增殖抑制活性,其中对人源急
性髓细胞白血病细胞系thp-1、kg-1、mv-4-11和hl-60的抑制活性最强,对骨髓瘤、淋巴瘤以及其他实体瘤细胞的抑制活性弱于人源急性髓细胞白血病细胞系(表3)。
[0141]
实施例17
[0142]
tp-p1在mv-4-11裸鼠移植瘤模型上的抗肿瘤效果
[0143]
实验方法:mv-4-11细胞体外培养扩增,取适量处于对数生长期的细胞重悬于无血清 imdm培养基与matrigel(1:1)混悬液中,无菌条件下制备成5
×
106/100μl细胞悬液,用注射器将100μl细胞悬液接种于雄性balb/c裸小鼠前左肢腋窝皮下;待肿瘤体积生长至 100-200mm3时,选取肿瘤大小适中的动物随机分组,每组5只;分别给予空白媒介(pbs)、受试化合物tp-p1低剂量(25μg/kg/d)、tp-p1中剂量(50μg/kg/d)、tp-p1高剂量(100 μg/kg/d),每天腹腔注射一次,给药4周;给药期间,每天测量裸小鼠体重和瘤径;实验结束后颈椎脱臼处死,取瘤称重。
[0144]
肿瘤体积(tumor volume,tv)的计算公式为:tv=1/2
×a×
b2,a表示肿瘤长径;b表示肿瘤短径。
[0145]
表4 tp-p1在mv-4-11裸鼠移植瘤模型上的抗肿瘤效果
[0146][0147]
*,p《0.05;**,p《0.01;***,p《0.001(与溶剂对照比较)。
[0148]
结果显示:在mv-4-11裸鼠移植瘤模型上,连续腹腔注射给药4周,化合物tp-p1能够剂量依赖性地抑制肿瘤的生长,并且对小鼠体重没有影响;其中25μg/kg/d剂量下的抑瘤率达到54.31%,100μg/kg/d剂量可使移植瘤完全消退,抑瘤率达到100%(表4)。
[0149]
实施例18
[0150]
tp-p1在thp-1裸鼠移植瘤模型上的抗肿瘤效果
[0151]
实验方法:thp-1细胞体外培养扩增,取适量处于对数生长期的细胞重悬于无血清1640 培养基与matrigel(1:1)混悬液中,无菌条件下制备成5
×
106/100μl细胞悬液,用注射器将 100μl细胞悬液接种于雄性balb/c裸小鼠前左肢腋窝皮下;待肿瘤体积生长至100-200mm3时,选取肿瘤大小适中的动物随机分组,每组5只;分别给予空白媒介(pbs)、阳性对照药tp(180μg/kg/d)、受试化合物tp-p1(100μg/kg/d)、tp-p1(300μg/kg/d)、tp-p1 (600μg/kg/d),tp-p1(1200μg/kg/d),每天腹腔注射一次,给药4周;给药期间,每天测量裸小鼠体重和瘤径。实验结束后颈椎脱臼处死,取瘤称重。
[0152]
肿瘤体积(tumor volume,tv)的计算公式为:tv=1/2
×a×
b2,a表示肿瘤长径;b表示肿瘤短径。
[0153]
表5 tp-p1在thp-1裸鼠移植瘤模型上的抗肿瘤效果
[0154][0155]
*,p《0.05;**,p《0.01;***,p《0.001(与溶剂对照比较)。
[0156]
结果显示:在thp-1裸鼠移植瘤模型上,连续腹腔注射给药4周,化合物tp-p1能够剂量依赖性地抑制肿瘤的生长,并且对小鼠体重没有影响。其中100μg/kg/d剂量下的抑瘤率达到93.87%,300μg/kg/d及以上剂量可使移植瘤完全消退,抑瘤率达到100%(表5)。
[0157]
实施例19
[0158]
tp-p1与吉瑞替尼联合给药在mv-4-11裸鼠移植瘤模型上的抗肿效果
[0159]
实验方法:mv-4-11细胞体外培养扩增,取适量处于对数生长期的细胞重悬于无血清 imdm培养基与matrigel(1:1)混悬液中,无菌条件下制备成5
×
106/100μl细胞悬液,用注射器将100μl细胞悬液接种于雄性balb/c裸小鼠前左肢腋窝皮下;待肿瘤体积生长至 100-200mm3时,选取肿瘤大小适中的动物随机分组,每组6只;分别给予空白媒介(pbs) 及羧甲基纤维素钠(cmc-na)、受试化合物tp-p1剂量组(50μg/kg/d)、吉瑞替尼低剂量(0.5mg/kg/d)、吉瑞替尼高剂量(1mg/kg/d),tp-p1联用吉瑞替尼低剂量组;每天 tp-p1腹腔注射,吉瑞替尼灌胃给药一次,给药3周;给药期间,每天测量裸小鼠体重和瘤径;实验结束后颈椎脱臼处死,取瘤称重。
[0160]
肿瘤体积(tumor volume,tv)的计算公式为:tv=1/2
×a×
b2,a表示肿瘤长径;b表示肿瘤短径。
[0161]
表6 tp-p1与吉瑞替尼联合给药在mv-4-11裸鼠移植瘤模型上的抗肿效果
[0162][0163]
*,p《0.05;**,p《0.01;***,p《0.001(与溶剂对照比较)。^,p《0.05;^^,p《0.01;^^^,p《 0.001(与吉瑞替尼低剂量组比较)。
[0164]
结果显示:在mv-4-11裸鼠移植瘤模型上,连续给药3周,联合组的抑瘤率78.12%高于对应剂量单药吉瑞替尼组(48.27%),且高于两倍剂量单药吉瑞替尼组(71.96%)(表6);因此,化合物tp-p1与吉瑞替尼联合用于急性髓细胞白血病的治疗具有协同增效作用。
[0165]
实施例20
[0166]
tp-p1缓解lps诱导的脓毒症小鼠肺部炎症的作用
[0167]
实验方法:将雌性balb/c小鼠采用随机分组法分为5组,每组6只,分别为溶剂对照组、模型组、tp-p1低剂量组(500μg/kg/d)、tp-p1中剂量组(1000μg/kg/d)、tp-p1高剂量组(1500μg/kg/d),各组均在注射lps前两天进行预防性给药,每天腹腔注射一次,连续给药2天,给药体积为10ml/kg;第3天,分别给予模型组、tp-p1低、中、高各剂量组腹腔注射一次10mg/kg lps,给药体积为5ml/kg;第4天,无菌摘取小鼠肺组织,通过 rt-qpcr技术检测il-1β、il-6、tnf-α、ifn-γ等炎性因子的表达情况,各组数据分别与溶剂对照组做归一化处理。
[0168]
表7 tp-p1缓解lps诱导的脓毒症小鼠肺部炎症的作用
[0169][0170][0171]
*,p《0.05;**,p《0.01;***,p《0.001(与溶剂对照组比较)。^,p《0.05;^^,p《0.01;^^^,p《 0.001(给药组与模型组比较)。
[0172]
结果显示:tp-p1能够抑制il-1β、il-6、tnf-α、ifn-γ等炎性因子的释放,并且呈
剂量依赖性,缓解lps诱导的脓毒症小鼠肺部炎症(表7)。
[0173]
实施例21
[0174]
tp-p1的药代动力学性质
[0175]
实验方法:将8只雄性sd大鼠采用随机分组法分为2组,随机分组的sd大鼠禁食过夜但可以自由饮水;12h后,第一组给予化合物tp-p1的水溶液,灌胃给药,给药剂量为1.6 mg/kg;第二组给予tp的橄榄油混悬液,灌胃给药,给药剂量为1.0mg/kg;采用眼眶取血法,分别于2min、5min、10min、15min、30min、45min、60min、90min、2h、4h、6 h取血,转移至肝素钠预先处理的1.5ml的离心管后离心分离(8000rpm/min,5min,4℃) 得到血浆,并保存于-80℃的冰箱中。
[0176]
处理方法:采用1:3甲醇蛋白沉淀法处理血浆样本,涡旋离心后,取上清通过 uplc-ms/ms分析血浆中的tp血药浓度,采用phoenix 64软件处理数据,得到药代动力学参数。
[0177]
表8 tp-p1的药代动力学性质
[0178][0179]
结果显示:tp-p1的auc
(0-t)
、cmax等药代参数都优于tp,在等摩尔口服给药的情况下tp-p1的吸收明显高于tp(表8)。
[0180]
以上仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1