重组杆状病毒基因组及其改造方法、应用以及表达外源蛋白的方法与流程
1.本发明涉及基因工程的技术领域,具体而言,涉及重组杆状病毒基因组及其改造方法、应用以及表达外源蛋白的方法。
背景技术:
2.杆状病毒属于双链dna病毒,其基因组大小为80-180kb,自然界仅感染节肢动物,主要感染昆虫纲的鳞翅目、膜翅目、双翅目以及甲壳纲的十足目。杆状病毒基因组主要特点是可同时容纳多个大片段基因并表达外源蛋白,通过昆虫细胞表达的外源蛋白可以得到很好的修饰,且具有良好的生物学活性,杆状病毒表达系统已被广泛应用于蛋白、疫苗等领域,被公认为是非常优良的表达系统。
3.但是,改善杆状病毒表达系统表达外源蛋白的产能问题一直是国内外学者的研究方向,杆状病毒表达系统所表达的外源蛋白虽然生物学活性高但是其产能问题一直是杆状病毒表达系统的瓶颈。acmnpv(苜蓿银纹夜蛾核型多角体病毒)是应用最广泛的杆状病毒,其基因组中的v-cath(组织蛋白酶)基因导致虫体液化促使病毒释放;chia(几丁质酶)基因的功能是促使虫体融化与v-cath有协同作用,与杆状病毒侵染虫体有关。v-cath/chia不影响病毒的正常复制,但是可能会降解表达的外源蛋白。p26-p10-p74基因为非必须基因,与病毒经口感染昆虫有关。vankrin(齿唇姬蜂病毒vankrin基因)属于抗凋亡基因,可以通过基因调控抑制细胞凋亡,ns1(家蚕浓核病毒ns1基因)参与病毒的复制,并可能参与病毒的装配,有利于病毒粒子的成熟。
4.red/et重组技术是新近出现的一种基于λ噬菌体red操纵子(redα/redβ/redγ)和rac噬菌体rece/rect重组系统的dna工程技术,是red同源重组系统和et同源重组系统的总称,结合rpsl-cmr反向筛选技术可以简单、快速地对任意大的dna分子进行插入、敲除、突变等多种修饰,而且还可对长达80kb的dna片段进行亚克隆。由于重组反应的整个过程都是在大肠杆菌细胞内部完成,因此不存在碱基突变危险。该技术已被广泛地用于基因组dna,如细菌人工染色体、大肠杆菌染色体等的遗传修饰研究以及基因工程药物的研究和开发中。
5.因此,使用基因重组技术剔除杆状病毒基因组中的某些非必须基因,并且相应的引入外源蛋白表达相关基因或许能够在一定程度上提高杆状病毒的外源蛋白表达水平。
技术实现要素:
6.本发明的目的在于提供了重组杆状病毒基因组的改造方法与之相适配的表达外源蛋白的方法,其不仅能够显著提高杆状病毒的增殖能力,还能够显著提高外源蛋白的表达水平。
7.本发明的实施例通过以下技术方案实现:本发明提供了一种重组杆状病毒基因组,命名为bac-δcp-vn,其生物保藏编号为:cgmcc no.23911,保藏地址为:北京市朝阳区北辰西路1号院3号,保藏日期:2021年11月15日。
8.本发明还提供了上述重组杆状病毒基因组的改造方法,包括使用基因重组系统和反向筛选系统敲除acmnpv中的chia、v-cath、p26、p10、p74基因,并在敲除位点分别引入vankrin和ns1基因,具体包括如下步骤:
9.(1)将bac-to-bac系统的病毒基因组中chia-v-cath以及p26-p10-p74基因敲除,命名为bac-δcp;
10.(2)在chia-v-cath基因的敲除位点引入vankrin(序列为seq id.no.1)基因,在p26-p10-p74基因的敲除位点引入ns1基因(序列为seq id.no.2),命名为bac-δcp-vn;
11.(3)将步骤(2)获得的bac-δcp-vn制备成感受态细胞。
12.进一步地,所述基因重组系统包括red/et重组系统和crispr/cas 9敲除系统中的一种;优选为red/et重组系统。
13.进一步地,所述反向筛选系统包括rpsl-cmr,hsv-tk、sacb、tetr反向筛选系统中的一种;优选为rpsl-cmr。
14.本发明还提供了基于上述重组杆状病毒基因组的表达外源蛋白的方法,包括如下步骤:
15.(1)构建包含外源基因的转移载体,pfastbac-gfp、pfastbac-cap、pfastbac-gc;其中转移载体为pfastbac;所述外源基因为gfp,pcv2-cap,prv-gc中的一种;其中gfp为绿色荧光蛋白,gfp的序列为seq id.no.3;pcv2-cap为猪圆环病毒的cap蛋白,pcv2-cap的序列为seq id.no.4;prv-gc为猪伪狂犬病毒的gc蛋白,prv-gc的序列为seq id.no.5。
16.(2)将步骤(1)所得的转移载体转化到bac-δcp-vn制备成感受态细胞中,获得重组bacmid质粒;
17.(3)将步骤(2)中获得的重组bacmid质粒转染昆虫细胞,获得重组杆状病毒;
18.(4)将步骤(3)获得的重组杆状病毒收获上清,进一步接种sf9细胞扩毒和接种hi5细胞表达检测其扩毒能力和病毒表达能力。
19.本发明还提供了上述重组杆状病毒基因组的改造方法应用于提高外源蛋白产能。
20.本发明实施例的技术方案至少具有如下优点和有益效果:本发明的重组杆状病毒基因组的改造方法,通过将acmnpv基因组中存在的chia、v-cath、p26、p10、p74非必需基因的敲除,同时在敲除位点引入vankrin与ns1基因,能够显著提高杆状病毒的增殖能力以及外源蛋白的表达水平。
附图说明
21.图1为本发明的实验例1的dh10bac-δcp-vn鉴定结果;
22.图2为本发明的实验例2的第8天荧光结果;
23.图3为本发明的实验例3的重组杆状病毒提取基因组pcr鉴定结果;
24.图4为本发明的实验例5的病毒滴度的统计结果;
25.图5为本发明的实验例6的7种重组毒株96h与192h表达结果;
26.图6为本发明的实施例6的7种重组毒株96h与192h表达结果统计。
具体实施方式
27.以下通过具体的实施例对本发明进一步阐述,本发明的3个实施例不对本发明的
范围构成任何限制。特别的,在本领域的技术人员在不偏离本发明主线的情况下,对本发明可以所做的优化与改进,但是这些优化和改进均落入本发明的保护范围内。
28.本发明实施例中所用到的化学试剂为分析纯,购自国药集团。
29.试剂:primerstar max和荧光定量pcr试剂(sybr green ii)购自takara宝生物工程(大连)有限公司。质粒提取试剂盒与胶回收试剂盒购自天根生物科技有限公司。bca蛋白浓度测定试剂盒购自biosharp公司。t5外切酶购自neb。碧云天杆状病毒穿梭载体bacmid小量抽提试剂盒。
30.本发明实施例中所用到的细胞培养基均为无血清培养基ib905,购自北京鼎持生物科技有限公司。
31.本发明实施例中所用到的引物或基因合成由擎科生物科技有限公司合成。
32.本发明所述的实验方法,若无特殊说明,均为常规方法。
33.实施例1
34.1 bac基因组5个基因敲除
35.1.1同源重组线性片段的准备
36.1.1.1同源重组线性片段updonor1-rpsl-cmr-downdonor1的准备
37.根据genbank上登录号为kf022001.1的acmnpv基因中同源重复序列chia上游区域和下游区域设计引物,根据rpsl-cmr序列设计上下游引物,通过pcr的方法分别扩增出updonor1、rpsl-cmr、downdonor1,overlap pcr形成打靶片段updonor1-rpsl-cmr-downdonor1,其序列如seq id.no.6,引物序列见表1,pcr反应条件见表2。updonor1与downdonor1模板均为未改造bacmid,rpsl-cmr(genbank no.nc_000913.3)由基因合成公司合成。
38.表1引物序列
[0039][0040]
表2 pcr反应条件
[0041][0042]
将扩增所得3个片段updonor1、rpsl-cmr、downdonor1,将3个核酸片段电泳并进行胶回收并进行overlappcr扩增得到完整的线性片段,模板以及反应条件见表3。
[0043]
表3overlap模板与反应条件
[0044][0045]
扩增完成后将pcr片段命名为updonor1-rpsl-cmr-downdonor1并胶回收并电泳检测,-20℃冻存。
[0046]
1.1.2同源重组线性片段updonor2-rpsl-cmr-downdonor2的准备
[0047]
根据genbank上登录号为kf022001.1的acmnpv基因中同源重复序列p26-p10-p74上游区域和下游区域设计引物,根据rpsl-cmr序列设计上下游引物,通过pcr的方法分别扩增出updonor2、rpsl-cmr、downdonor2,overlap pcr形成打靶片段updonor2-rpsl-cmr-downdonor2,其序列如seq id.no.7,引物序列见表4,pcr反应条件见表2。updonor2与downdonor2模板均为未改造bacmid,rpsl-cmr(genbank no.nc_000913.3)由基因合成公司合成。
[0048]
表4引物序列
[0049]
[0050][0051]
将扩增所得3个片段updonor2、rpsl-cmr、downdonor2将3个核酸片段电泳并进行胶回收,再进行overlappcr扩增,将3个片段融合成一个完整的线性片段,模板以及反应条件见表5。
[0052]
表5 overlap模板与反应条件
[0053][0054]
扩增完成后将pcr片段命名为upd-rpsl-cmr-downd并胶回收并电泳检测,-20℃冻存。
[0055]
1.2含pkd46 dh10bac电转感受态制备
[0056]
1.2.1 dh10bac电转感受态制备
[0057]
将dh10bac菌株平板划线于lb(含kana、tet)固体平板,37℃培养静置培养过夜。挑取单克隆菌在lb(含kana、tet)液体培养基中活化,37℃220rpm培养过夜。菌种活化后按1:100转接至lb(含kana、tet)培养基中,37℃220rpm培养到od为0.4-0.6,取出后冰浴10min。4℃4000rpm离心5min,轻轻弃去上清液。使用无菌dd h2o连续洗两次。最后将菌团使用1ml无菌预冷的10%甘油重悬,按100μl/管分装,保存于-80℃冰箱备用。
[0058]
1.2.2电转化pkd46质粒
[0059]
将pkd46质粒加入到融化的dh10bac感受态中轻轻混匀转移至电转杯中,调节电转仪的电压为1.8kv,放入电转仪,电转化时间为4-6ms,电转后向电转杯中加入1ml的lb培养基(不含抗生素),30℃摇床220rpm培养60min涂布于lb(含kana、tet、amp)固体平板中,30℃培养箱培养36h,命名为dh10bac-pkd46。
[0060]
1.2.3 dh10bac-pkd46电转感受态制备
[0061]
挑取实施例1中2.2获得的dh10bac-pkd46单克隆菌在lb(含kana、tet、amp)液体培养基中活化,30℃220rpm培养过夜。菌种活化后按1:100转接至lb(含kana、tet、amp、l-ara)
培养基中,30℃220rpm培养到od为0.4-0.6,取出后冰浴30min。离心后轻轻弃去上清液。使用无菌ddh2o联系洗两次。最后将菌团使用1ml无菌预冷的10%甘油重悬,按100μl/管分装,保存于-80冰箱备用。
[0062]
1.3电转化同源重组线性片段
[0063]
将实施例1中1.1获得的updonor1-rpsl-cmr-downdonor1以及1.2获得的updonor2-rpsl-cmr-downdonor2加入到实施例1中2.3获得的dh10bac-pkd46电转感受态中,轻轻混匀后转移至电转杯中,调节电转仪的电压为1.8kv,电转化时间设定为4-6ms。上向电转杯中加入1ml的lb培养基(不含抗生素),30℃摇床220rpm培养60min涂布于lb(含kana、tet、amp、cm)固体平板中,30℃培养箱培养36h,命名为dh10bac-pkd46/δcp。
[0064]
1.4pcr验证
[0065]
将实施例1中第3项所得dh10bac-pkd46/δcp利用鉴定引物进行pcr扩增。引物序列见表5,反应条件见表6。
[0066]
表5引物序列
[0067][0068]
表6 pcr反应条件
[0069][0070]
1.5 dh10bac-pkd46/δcp电转感受态制备
[0071]
将实施例1中第4项获得的验证正确的dh10bac-pkd46/δcp取单克隆活化,30℃220rpm培养过夜。菌种活化后按1:100转接至lb(含kana、tet、amp、l-ara)培养基中,30℃220rpm培养到od为0.4-0.6,取出后冰浴30min,离心后轻轻弃去上清液。使用无菌ddh2o连续洗两次。最后将菌团使用1ml无菌预冷的10%甘油重悬,按100μl/管分装,保存于-80冰箱备用。
[0072]
1.6 pkd46消除与验证
[0073]
实施例1第4项验证正确的dh10bac-pkd46/δcp,lb(kan+cm+tet)摇菌,37℃培养过夜。取100μl菌液10-6稀释后涂板(kan+cm+tet),37℃培养过夜,从过夜培养的平板中挑20个单克隆分别接种至lb+amp+kan+cm和lb+kan+cm固体平板上。若某个克隆在lb+kan+cm
培养基中正常生长,而在lb+amp+kan+cm中不生长,则说明pkd46被消除,正确菌株命名为dh10bac-δcp。
[0074]
2 dh10bac-δcp中引入vankrin与ns1基因
[0075]
2.1插入同源重组基因片段准备
[0076]
2.1.1 updonor3-vankrin-downdonor3与updonor4-ns1-downdonor4线性片段制备
[0077]
以改造前bacmid为模板,通过pcr的方法分别扩增出updonor3、downdonor3、updonor4、downdonor4,以合成的vankrin-ns1基因为模板扩增出vankrin表达核(seq id.no.1)和ns1表达核(seq id.no.2),引物序列见表7,pcr反应条件见表8。
[0078]
表7引物序列
[0079]
updonor3-vankrin-downdonor3相关基因片段扩增
[0080][0081][0082]
updonor4-ns1-downdonor4相关基因片段扩增
[0083][0084]
表8 pcr反应条件
[0085][0086]
将3个核酸片段电泳并进行胶回收,再进行overlappcr扩增,将3个片段融合成一个完整的线性片段,模板以及反应条件见表9。
[0087]
表9 overlap模板与反应条件
[0088][0089]
扩增完成后将pcr片段分别命名为updonor3-vankrin-downdonor3、updonor4-ns1-downdonor4,-20℃冻存。
[0090]
2.2电转化同源重组片段
[0091]
将步骤2.1.1中所获得的updonor3-vankrin-downdonor3、updonor4-ns1-downdonor4两个同源重组片段加入到实施例1第5项获得的dh10bac-pkd46/δcp感受态中,混合后将混合物转移至电转杯中,电转压力位1.8kv,时间为4-6ms。向电转杯中加入1ml的lb培养基(无抗生素),30℃摇床220rpm培养60min涂布于lb(含kana、tet、amp、cm、str)固体平板中,30℃培养箱培养36h,命名为dh10bac-pkd46/δcp-vn。
[0092]
2.3 pkd46质粒的消除与验证
[0093]
将步骤2.2验证正确的dh10bac-pkd46/δcp-vn,lb(kan+cm+tet)摇菌,37℃培养过夜。取100μl菌液10-6稀释后涂板(kan+cm+tet),37℃培养过夜,从过夜培养的平板中挑20个单克隆分别接种至lb+amp+kan+cm和lb+kan+cm固体平板上。若某个克隆在lb+kan+cm培养基中正常生长,而在lb+amp+kan+cm中不生长,则说明pkd46被消除,正确菌株命名为dh10bac-δcp-vn。
[0094]
3转移载体构建
[0095]
3.1插入片段gfp,pcv2-cap,prv-gc基因的获得。
[0096]
以合成的seq id.no.3,seq id.no.4,seq id.no.5为模板,扩增含有骨架同源臂(20bp)的pcr-gfp,pcr-pcv2-cap,pcr-prv-gc,引物序列见表10,pcr扩增条件见表11。
[0097]
表10引物序列
[0098][0099]
表11 pcr反应条件
[0100][0101][0102]
3.2供体质粒的获得
[0103]
在商品化的pfastbac载体的ph启动子后插入hbm信号肽作为供体质粒,通过ecor i和kpni酶切2μl,37℃酶切2h,胶回收载体骨架,并核酸电泳,命名为pfastbac-hbm-ecor i/kpn i。
[0104]
3.3热转感受态制备
[0105]
将ez10菌株平板划线于lb固体平板,37℃培养静置培养过夜。挑取单克隆菌在lb液体培养基中活化,37℃220rpm培养过夜。按1:100转接37℃220rpm培养到od为0.4-0.6,取出后冰浴30min后4℃4000rpm离心5min,轻轻弃去上清液。无菌0.1mcacl2洗两次。最后将菌团使用10ml无菌预冷的重悬液(含0.1mol/l的cacl2+16.7%甘油)重悬,按100μl/管分装,保存于-80冰箱备用。
[0106]
3.4重组转移载体构建
[0107]
将步骤3.1中获得的pcr-gfp,pcr-pcv2-cap,pcr-prv-gc分别与步骤3.2获得的供体质粒pfastbac-hbm-ecor i/kpni进行t5克隆,完成后直接加入到步骤3.3获得的ez10感受态中,冰浴30min,42℃水浴90s,再次冰浴3min,向ep管中加入500μl无抗lb培养基,37℃220rpm培养60min后涂lb(amp)固体培养板,37℃静置过夜,分别命名为pfastbac-gfp、
pfastbac-cap、pfastbac-gc。
[0108]
挑取菌落进行pcr鉴定,每个菌板挑取5个单克隆,分别使用10μl lb重悬后取1μl进行pcr鉴定,引物同表10,pcr鉴定同表11。阳性菌落测序。
[0109]
4重组bacmid的获得
[0110]
4.1取步骤3.4所得的pfastbac-gfp取1μl加入到步骤1.6获得的dh10bac-δcp、步骤2.3获得的dh10bac-δcp-vn以及未改造的dh10bac感受态细胞中,轻弹混匀,冰上孵育30min,42℃热激90s,冰上孵育3min后加入500μl的lb培养基37℃220rpm 60min,取100μl lb重悬菌液涂布于含有iptg/x-gal/kana/tet/gm三抗平板,37℃培养过夜,当可观察到明显的蓝白斑时,挑取白斑进行pcr鉴定,鉴定引物为m13f与gm-r序列如表12所示,pcr扩增条件同表11。
[0111]
表12引物序列
[0112][0113]
将阳性重组bacmid分别命名为dh10bac-δcp-vn-gfp、dh10bac-δcp-gfp、dh10bac-gfp。
[0114]
4.2取实施例3第4项所得的pfastbac-cap、pfastbac-gc各取1μl分别加入到实施例2第3项获得的dh10bac-δcp-vn以及未改造的dh10bac感受态细胞中,冰上孵育30min,42℃热激90s,冰上孵育3min后加入500μl的lb培养基37℃220rpm 60min,取100μl lb重悬菌液涂布于含有iptg/x-gal/kana/tet/gm三抗平板,37℃培养过夜,当可观察到明显的蓝白斑时,挑取白斑进行pcr鉴定,鉴定引物如表12所示,pcr扩增条件同表11。
[0115]
将阳性重组bacmid分别命名为dh10bac-δcp-vn-cap、dh10bac-δcp-vn-gc、dh10bac-cap、dh10bac-gc。
[0116]
4.3重组杆状病毒基因组获得
[0117]
将步骤4.1和步骤4.2验证正确的菌株dh10bac-δcp-gfp、dh10bac-δcp-vn-gfp、dh10bac-gfp、dh10bac-δcp-vn-cap、dh10bac-δcp-vn-gc、dh10bac-cap、dh10bac-gc转接至10ml lb(kana/tet/gm)液体培养基中,37℃220rpm培养过夜,使用杆状病毒穿梭载体bacmid小量抽提试剂盒对重组杆粒进行提取,所得bacmid分别命名为bac-δcp-gfp、bac-δcp-vn-gfp、bac-gfp、bac-δcp-vn-cap、bac-δcp-vn-gc、bac-cap、bac-gc。
[0118]
4.4重组杆状病毒的获得及扩大培养并鉴定
[0119]
将步骤4.3获得的重组bac基因组bac-δcp-gfp、bac-δcp-vn-gfp、bac-gfp、bac-δcp-vn-cap、bac-δcp-vn-gc、bac-cap、bac-gc分别转染至sf9细胞中,并观察荧光情况。6d后收获培养基上清,并分别命名为p1-bac-δcp-gfp、p1-bac-δcp-vn-gfp、p1-bac-gfp、p1-bac-δcp-vn-cap、p1-bac-δcp-vn-gc、p1-bac-cap、p1-bac-gc。
[0120]
p1代毒按1%盲传至p2代,并命名为p2-bac-δcp-gfp、p2-bac-δcp-vn-gfp、p2-bac-gfp、p2-bac-δcp-vn-cap、p2-bac-δcp-vn-gc、p2-bac-cap、p2-bac-gc,第4d收毒,向含gfp 3个毒株的t25方瓶补充培养基,继续放置至8d。
[0121]
实验例1
[0122]
dh10bac-δcp-vn鉴定
[0123]
实施例的步骤2.2所获得的每个菌落,将每个菌落使用两套引物进行扩增,若均正确则打靶成功,其余均为错误打靶。引物见表13,结果如图1所示。附图1a为vankrin替换chia-v-cath基因验证,1~10为挑取的10个单克隆菌,nc为阴性对照,pc为阳性对照。图1b为ns1替换p26-p10-p74基因验证,1~10为挑取的10个单克隆菌,nc为阴性对照,pc为阳性对照。由附图1a和附图1b可以看出,vankrin成功替换了chia-v-cath;ns1成功替换了p26-p10-p74。
[0124]
表13打靶鉴定引物
[0125][0126]
实验例2
[0127]
细胞表面荧光检测
[0128]
将实施例的步骤4.4中所得到的放置8d后的病毒进行细胞表面荧光观察。结果如附图2所示,附图2中,a:p2-bac-gfp、b:p2-bac-δcp-gfp、c:p2-bac-δcp-vn-gfp;可以看出,放置第8天时,非必须基因敲除的重组病毒荧光量明显高于非改造的重组病毒,但荧光量均未明显增加,而有vankrin-ns1基因的重组病毒荧光蛋白不断富集,说明敲除非必须基因后,有利于蛋白的表达,同时插入vankrin-ns1基因后,有利于蛋白表达的进一步提高。
[0129]
实验例3
[0130]
重组杆状病毒构建检测
[0131]
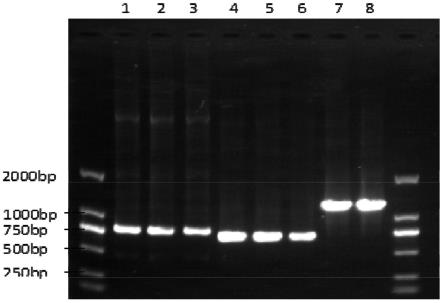
将实施例的步骤4.4中所得到的p2代病毒使用天根病毒dna提取试剂盒提取基因组,使用不同蛋白对应的特异性引物扩增,引物同实施例中的步骤3.1中的表11,结果如图3所示,附图3中,1、p3-bac-δcp-gfp;2、p3-bac-δcp-vn-gfp;3、p3-bac-gfp;4、p3-bac-δcp-vn-cap;5、p2-bac-cap;6、p3-bac-cap;7、p3-bac-δcp-vn-gc;8、p3-bac-gc。由附图3可以看出,7种重组杆状病毒种均插入相应的目的基因,重组杆状病毒构建成功。
[0132]
实验例4
[0133]
病毒滴度测定
[0134]
对实施例的步骤4.4中所获得的p2-bac-δcp-gfp、p2-bac-δcp-vn-gfp、p2-bac-gfp进行病毒滴度测定。铺96孔板,按1
×
105cell/孔,100μl/孔,27℃静置培养。使用1.5ml无菌ep管将3种毒株分别进行10倍倍比稀释,从10
1-109,将稀释好的病毒接种到96孔培养板中,每一稀释度接种一纵排,共8孔,每孔接种100μl。每日显微镜下观察荧光孔数并记录结果,一般需要观察5天,按reed-muench两氏法计算病毒滴度。检测结果如表14所示。
[0135]
对实施例的步骤4.4中所获得的p2-bac-δcp-vn-cap、p2-bac-δcp-vn-gc、p2-bac-cap、p2-bac-gc进行病毒滴度测定。病毒稀释并接种到96孔培养板中。27℃培养5天后,
使用丙酮固定液固定,37℃孵育gp64单抗60分钟。洗板后孵育带fitc荧光标记二抗,观察:在荧光显微镜下观察并记录出现荧光的孔数,按reed-muench两氏法计算病毒tcid50。检测结果如表14所示。
[0136]
表14不同重组杆状病毒tcid
50
[0137] 病毒滴度(第6d)p2-bac-gfp10
6.7
tcid50/mlp2-bac-δcp-gfp10
7.9
tcid50/mlp2-bac-δcp-vn-gfp10
8.70
tcid50/mlp2-bac-cap10
7.2
tcid50/mlp2-bac-δcp-vn-cap10
8.5
tcid50/mlp2-bac-gc10
7.1
tcid50/mlp2-bac-δcp-vn-gc10
8.75
tcid50/ml
[0138]
从表14的病毒滴度结果可以看出,将chia-v-cath,p26-p10-p74基因敲除可提高病毒滴度,插入vankrin与ns1基因后进一步提高了病毒滴度。表明本方法改造的bac-δcp-vn可以明显的提高病毒滴度。
[0139]
实验例5
[0140]
病毒扩增
[0141]
将实施例的步骤4.4中所得到的p2代重组杆状病毒p2-bac-δcp-gfp、p2-bac-δcp-vn-gfp、p2-bac-gfp、p2-bac-δcp-vn-cap、p2-bac-δcp-vn-gc、p2-bac-cap、p2-bac-gc按0.1moi分别接种sf9细胞(125摇瓶),分别于4d、6d、8天收获培养基上清,检测病毒滴度。方法同实验例3。结果如表15所示。
[0142]
表15重组杆状病毒病毒滴度测定结果
[0143][0144][0145]
从表15的病毒滴度测定结果可以看出不同毒株均按0.1moi接毒后,未改造抗原病毒滴度始终较低107tcid50/ml左右,而敲除5个非必须基因后,病毒滴度有所提高,但是从第4日开始病毒滴度不再增加,当插入vankrin和ns1基因后,病毒滴度不断增加,相比对未改造bac-to-bac系统病毒滴度增加了100倍(如图4所示)。说明插入的两种基因起到了明显的增加病毒滴度的作用。
[0146]
实验例6
[0147]
蛋白的表达
[0148]
将实施例的步骤4.4中获得的p2代重组杆状病毒p2-bac-δcp-gfp、p2-bac-δcp-vn-gfp、p2-bac-gfp、p2-bac-δcp-vn-cap、p2-bac-δcp-vn-gc、p2-bac-cap、p2-bac-gc按1moi分别接种hi5细胞,分别于96h和192h收取样品并电泳检测。电泳以及蛋白定量结果如附图5和附图6所示。
[0149]
由附图5-附图6可以看出,72h和192h所收获的样品检测结果一致,bac-δcp-vn》bac-δcp》bac;bac-δcp的蛋白表达量明显高于bac,但是两种基因组表达的目的蛋白均在第4天以后不再增加,而bac-δcp-vn表达的蛋白对时间的推移蛋白含量不断增加,证明了敲除5个非必须基因后增加了目的蛋白的表达,同时插入vankrin与ns1基因后促使目的蛋白持续表达,产能持续增加。
[0150]
综上,从实施例以及实验例1-6可以看出,本发明的改造方法表明不仅成功将vankrin替换了chia-v-cath,ns1替换了p26-p10-p74,成功构建了重组杆状病毒,并且病毒滴度检测结果也表明插入vankrin与ns1基因后进一步提高了病毒滴度。蛋白表达检测也表明证明了敲除5个非必须基因后增加了目的蛋白的表达,同时插入vankrin与ns1基因后促使目的蛋白持续表达,产能持续增加。这为提高外源蛋白表达提供了新的思路与方向。
[0151]
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1