一种刷状核酸组装体和复合纳米粒子及其应用的制作方法
1.本发明涉及医药技术领域,具体涉及一种刷状核酸组装体和复合纳米粒子及其应用。
背景技术:
2.近年来,寡核苷酸的生产、纯化和细胞内递送等方面的研究进展迅猛,使得基于寡核苷酸的治疗方法得到广泛的应用。随着相关药品种类迅速扩大,基于寡核苷酸的基因疗法或将改变许多疾病的治疗标准并达到个性化医疗水平。此类药物具有成本效益,生产相对简单,并且可靶向此前不可成药的靶点和通路,是一种颠覆性的治疗技术。尤其小型生物科技初创企业和学术团体可以快速开发新的个性化构建的药物体系,为基因疗法的可持续发展注入新鲜活力。
3.寡核苷酸药物通常通过识别内源性rna转录本的互补序列并与之杂交,并改变其加工过程,诱导基因沉默。其中包括反义寡核苷酸(aso)和利用rna干扰技术(rnai)的寡核苷酸rna。aso通常为长度大约20个碱基的单链寡核苷酸,能与其靶向的mrna结合以沉默相应的基因表达。当aso通过碱基配对作用与靶向mrna结合时,rna/dna杂交双链作为核糖核酸酶(rnase h)的底物被降解,进而导致靶向mrna的降解。此外,aso还可以通过序列互补结合靶向rna并在不诱导其降解的情况下阻断mrna的翻译或调节mrna的剪接,从而达到基因调控的目的。与aso类似,基于rna干扰技术的治疗性rna小核酸药物可以通过先天性的生物过程抑制基因表达。真核生物通过产生内源性的微小rna(mirna)与胞质中的argonaute蛋白结合形成rna诱导的沉默复合物(risc)来调节mrna的翻译。risc复合物可以抑制mrna的翻译或促进mrna的降解。同样,小干扰rna(small interference rna,sirna)是外源性的双链rna片段(大多长度为15~30个碱基对),可以利用risc复合物结合并切断特定序列的mrna,以抑制翻译。改进的sirna结构克服了最初在细胞毒性和基因沉默效率方面的缺陷,近年来多款小核酸药物获得了监管机构的批准,重新树立了其作为治疗性药物的地位。
4.尽管寡核苷酸药物的研究取得了很大的进步,但药物递送问题仍然是其临床使用中面临的主要障碍。病毒载体具有与生俱来的能力可以将遗传物质递送至细胞内,是研究最广泛的基因载体。然而,病毒载体在临床转化中存在诸多安全问题(如免疫原性、潜在的毒副作用和插入突变)以及其他障碍(如复杂的制造过程、有限的负载容量)。
5.非病毒递送体系通常比病毒载体效率低,但更安全,更经济,免疫原性更低,并能携带更大的基因药物。其中,聚合物是常用的非病毒递送体系。例如,聚赖氨酸(pll)、聚乙烯亚胺(pei)以及聚酰胺-胺(pamams)树状分子在基因递送中应用较为广泛,但上述聚合物通常存在较强的细胞毒性,必须经过化学修饰以解决递送毒性问题。例如,中国专利cn107281497a公开了一种基于dna水凝胶的功能性核酸保护性载体,由包括侧链修饰dna的可降解高分子化合物、功能性核酸和交联剂自组装所得。然而,上述基于dna水凝胶的功能性核酸保护性载体对寡核苷酸药物的递送效率低。
技术实现要素:
6.有鉴于此,本发明的目的在于提供一种刷状核酸组装体和复合纳米粒子及其应用,本发明提供的侧链修饰dna的可降解高分子化合物能够实现对寡核苷酸药的有效压缩,对其递送效率高。
7.为了实现上述发明目的,本发明提供以下技术方案:
8.本发明提供了一种刷状核酸组装体,包含dna/rna杂交双链,由侧链修饰dna的可降解高分子化合物和具有单链rna粘性末端的功能性核酸分子经碱基互补组装得到。
9.优选的,所述侧链修饰dna的可降解高分子化合物由侧链修饰r1基的可降解高分子与端基修饰r2基的dna通过点击化学反应得到;
10.当所述r1基为叠氮基时,r2基包括炔基或二苯基环辛炔基;
11.当所述r1基为炔基或二苯基环辛炔基时,r2基包括叠氮基团;
12.当所述r1基为巯基时,r2基包括马来酰亚胺基;
13.当所述r1基为马来酰亚胺基时,r2基包括巯基。
14.优选的,所述侧链修饰dna的可降解高分子化合物中的dna的碱基数≥8。
15.优选的,所述侧链修饰dna的可降解高分子化合物中的可降解高分子化合物包括聚己内酯、聚乳酸、聚乳酸-羟基乙酸共聚物、聚β-氨基酯或聚肽。
16.优选的,所述具有单链rna粘性末端的功能性核酸分子包括rna粘性末端和功能性核酸分子,所述功能性核酸分子包括反义寡核苷酸、小干扰核酸、微小核糖核酸。
17.优选的,所述侧链修饰dna的可降解高分子化合物中的dna与具有单链rna粘性末端的功能性核酸分子中的单链rna粘性末端的摩尔比为1:0.1~1。
18.本发明提供了一种复合纳米粒子,由上述技术方案所述的刷状核酸组装体与阳离子聚合物自组装得到。
19.优选的,所述阳离子聚合物包括聚β-氨基酯、聚氨基酯、聚乙烯亚胺、聚赖氨酸、聚精氨酸、聚酰胺-胺和壳聚糖中的一种或几种。
20.优选的,所述刷状核酸组装体中核酸与阳离子聚合物的质量比为1:10~100。
21.本发明提供了上述技术方案所述的刷状核酸组装体或上述技术方案所述的复合纳米粒子在非疾病诊断或治疗中基因调控或制备核酸药物中的应用。
22.本发明提供了一种刷状核酸组装体,包含dna/rna杂交双链,由侧链修饰dna的可降解高分子化合物和具有单链rna粘性末端的功能性核酸分子经碱基互补组装得到。与现有技术相比,本发明通过将难以有效压缩的寡核苷酸药物等功能核酸分子设计为携带单链rna粘性末端的功能性核酸分子,然后与侧链修饰dna的可降解高分子化合物经过碱基互补形成拓扑结构大的刷状核酸组装体,提高了功能性核酸分子的负载率,提高其与阳离子聚合物的相互作用,有利于其与阳离子聚合物形成复合纳米粒子,进而提高了寡核苷酸药物等功能核酸分子的递送率。而且,刷状核酸组装体中dna/rna杂交双链部分能够被核酸酶rnase h识别和降解,从而释放出功能核酸部分,实现了高效核酸递送和基因调控效果。本发明提供的刷状核酸组装体为非病毒递送体系,免疫原性低,安全性高,成本低。
23.本发明提供了一种复合纳米粒子,由上述技术方案所述的刷状核酸组装体与阳离子聚合物自组装得到。在本发明中,刷状核酸组装体与阳离子聚合物自组装形成复合纳米粒子,能够富集功能性核酸并形成高电荷密度的多价拓扑核酸结构,从而实现了在使用低
剂量阳离子载体条件下高效压缩功能核酸分子并形成稳定的核酸/聚合物复合纳米粒子用于功能核酸的递送,毒性低,成本低。本发明提供的复合纳米粒子在生理条件下具有良好的稳定性,能够有效保护复合纳米粒子中所负载的功能核酸,避免被环境中核酸酶降解,能够高效地将所负载功能核酸递送至组织和细胞中。依靠复合纳米粒子中阳离子聚合物的“质子海绵”效应促进功能性核酸从内含体逃逸,从而将功能核酸高效递送至细胞质中,使得复合纳米粒子具有良好的基因调控功能。
附图说明
24.图1为实施例1中聚合物2/刷状核酸组装体a复合纳米粒子的制备路线示意图;
25.图2为实施例1制备的聚合物1(dna-g-plc)、刷状核酸组装体a(siegfp-brush)以及聚合物2/刷状核酸组装体a复合纳米粒子的水合粒径数据图;
26.图3为实施例1制备的中间产物3的1h nmr谱图;
27.图4为实施例1制备的聚合物2的1h nmr谱图;
28.图5为实施例1制备的中间产物3和聚合物2的凝胶渗透色谱数据图;
29.图6为带粘性末端靶向egfp的干扰小核酸(siegfp)、实施例1制备的聚合物1(dna-g-plc)和刷状核酸组装体a(siegfp-brush)的2%琼脂糖电泳图;
30.图7为实施例1制备的刷状核酸组装体a(siegfp-brush)的透射电子显微镜图;
31.图8为带粘性末端靶向egfp的干扰小核酸(siegfp)、刷状核酸组装体a(siegfp-brush)和实施例1制备的聚合物2/刷状核酸组装体a复合纳米粒子的0.5%琼脂糖电泳图;
32.图9为带粘性末端靶向egfp的干扰小核酸(siegfp)和对比例1制备的聚合物2/siegfp复合物的0.5%琼脂糖电泳图;
33.图10为实施例1制备的聚合物2/刷状核酸组装体a复合纳米粒子的透射电子显微镜图;
34.图11为带粘性末端靶向egfp的干扰小核酸(siegfp)、刷状核酸组装体a(siegfp-brush)、实施例1制备的聚合物1(dna-g-plc)以及聚合物2/刷状核酸组装体a复合纳米粒子与含10%fbs的dmem培养基孵育不同时间的0.5%琼脂糖电泳图;
35.图12为带粘性末端靶向egfp的干扰小核酸(siegfp)和实施例1制备的聚合物2/刷状核酸组装体a复合纳米粒子在不同rnasea酶浓度条件下孵育后的10%变性凝胶电泳图;
36.图13为实施例1制备的聚合物2/刷状核酸组装体a复合纳米粒子和对比例1制备的聚合物2/siegfp复合物的转染持续表达增强型绿色荧光蛋白的293t细胞后的细胞荧光流式分析图;
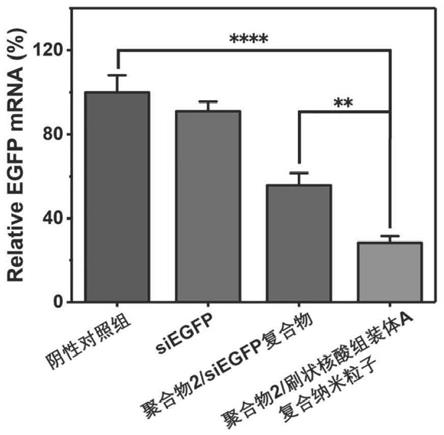
37.图14为带粘性末端靶向egfp的干扰小核酸(siegfp)、对比例1制备的聚合物2/siegfp复合物和实施例1制备的聚合物2/刷状核酸组装体a复合纳米粒子转染持续表达增强型绿色荧光蛋白的293t细胞后的实时荧光定量pcr数据图;
38.图15为实施例2制备的聚合物2/刷状核酸组装体b复合纳米粒子的制备路线示意图;
39.图16为带粘性末端靶向tgfβ1的干扰小核酸(sitgfβ1)、聚合物1(dna-g-plc)和实施例2制备的刷状核酸组装体b的2%琼脂糖电泳图;
40.图17为实施例2制备的刷状核酸组装体b(sitgfβ1-brush)和聚合物2/刷状核酸组
装体b复合纳米粒子的水合粒径结果;
41.图18为实施例2制备的刷状核酸组装体b(sitgfβ1-brush)的透射电子显微镜图;
42.图19为实施例2制备的聚合物2/刷状核酸组装体b复合纳米粒子的0.5%琼脂糖电泳图;
43.图20为对比例2制备的聚合物2/sitgfβ1复合物的0.5%琼脂糖电泳图;
44.图21为实施例2制备的聚合物2/刷状核酸组装体b复合纳米粒子的透射电子显微镜图;
45.图22为带粘性末端靶向tgfβ1的干扰小核酸(sitgfβ1)、对比例2制备的聚合物2/sitgfβ1复合物和实施例2制备的聚合物2/刷状核酸组装体b复合纳米粒子转染增生性瘢痕细胞后的实时荧光定量pcr数据图;
46.图23为带粘性末端靶向tgfβ1的干扰小核酸(sitgfβ1)、对比例2制备的聚合物2/sitgfβ1复合物和实施例2制备的聚合物2/刷状核酸组装体b复合纳米粒子对增生性瘢痕细胞周期抑制作用的示意图;
47.图24为实施例3制备聚合物3/刷状核酸组装体a复合纳米粒子的制备路线示意图;
48.图25为实施例3制备的中间产物4的1h nmr谱图;
49.图26为实施例3制备的聚合物3的1h nmr谱图;
50.图27为实施例3制备的聚合物3的凝胶渗透色谱数据图;
51.图28为带粘性末端靶向egfp的干扰小核酸(siegfp)、刷状核酸组装体a(siegfp-brush)、实施例3制备的聚合物3/刷状核酸组装体a复合纳米粒子的0.5%琼脂糖电泳图;
52.图29为实施例3制备的聚合物3/刷状核酸组装体a复合纳米粒子的透射电子显微镜图;
53.图30为带粘性末端靶向egfp的干扰小核酸(siegfp)、对比例3制备的聚合物3/siegfp复合物和实施例3制备的聚合物3/刷状核酸组装体a复合纳米粒子转染持续表达增强型绿色荧光蛋白的293t细胞后的细胞荧光流式分析图;
54.图31为实施例4制备的聚合物4/刷状核酸组装体a复合纳米粒子的制备路线示意图;
55.图32为带粘性末端靶向egfp的干扰小核酸(siegfp)、刷状核酸组装体a(siegfp-brush)、实施例4制备的聚合物4/刷状核酸组装体a复合纳米粒子的0.5%琼脂糖电泳图;
56.图33为实施例4制备的步骤1制备的聚合物4/刷状核酸组装体a复合纳米粒子水合粒径结果;
57.图34为实施例4制备的聚合物4/刷状核酸组装体a复合纳米粒子的透射电子显微镜图;
58.图35为带粘性末端靶向egfp的干扰小核酸(siegfp)、对比例4制备的聚合物4/siegfp复合物和实施例4制备的聚合物4/刷状核酸组装体a复合纳米粒子转染持续表达增强型绿色荧光蛋白的293t细胞后的细胞荧光流式分析图。
具体实施方式
59.本发明提供了一种刷状核酸组装体,包含dna/rna杂交双链,由侧链修饰dna的可降解高分子化合物和具有单链rna粘性末端的功能性核酸分子经碱基互补组装得到。
60.在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
61.在本发明中,所述具有单链rna粘性末端的功能性核酸分子中的功能性核酸分子中的单链rna粘性末端与所述侧链修饰dna的可降解高分子化合物中的dna碱基互补。在本发明中,所述互补的碱基数优选≥8,更优选为8~50,进一步优选为20~30。在本发明中,所述侧链修饰dna的可降解高分子化合物中的dna与具有单链rna粘性末端的功能性核酸分子中的单链rna粘性末端的摩尔比优选为1:0.1~1,更优选为1:0.2~0.8,进一步优选为1:0.4~0.5。
62.在本发明中,所述侧链修饰dna的可降解高分子化合物优选由侧链修饰r1基团的可降解高分子与端基修饰r2基团的dna通过点击化学反应得到。在本发明中,当所述r1基团为叠氮基团时,r2基团优选包括炔基或二苯基环辛炔(dbco)基;当所述r1基团为炔基或二苯基环辛炔基时,r2基团优选包括叠氮基团;当所述r1基团为巯基时,r2基团优选包括马来酰亚胺基团;当所述r1基团为马来酰亚胺基团时,r2基团优选包括巯基。在本发明中,所述侧链修饰dna的可降解高分子化合物中的dna的碱基数优选≥8,更优选为8~50,进一步优选为20~30。在本发明中,所述可降解高分子化合物优选包括聚己内酯、聚乳酸、聚乳酸-羟基乙酸共聚物、聚β-氨基酯或聚肽。
63.在本发明中,所述侧链修饰dna的可降解高分子化合物的制备方法,优选包括以下步骤:将侧链修饰r1基团的可降解高分子与端基修饰r2基团的dna混合,进行点击化学反应,得到侧链修饰dna的可降解高分子化合物。
64.在本发明中,所述端基修饰r2基团的dna优选委托生工生物工程(上海)股份有限公司合成。
65.本发明对于所述侧链修饰r1基团的可降解高分子的制备方法没有特殊限定,采用本领域技术人员熟知的制备方法即可,具体如先对可降解高分子化合物进行侧链修饰,得到侧链修饰r3基团的可降解高分子,然后将所述侧链修饰r3基团的可降解高分子通过缩合反应、取代反应或加成反应得到侧链修饰r1基团的可降解高分子,其中,r3基团优选包括羟基、氨基、羧基或卤素原子。
66.在本发明中,所述侧链修饰r1基团的可降解高分子与端基修饰r2基团的dna进行点击反应时两者比例优选为r1与r2基团摩尔比范围1:1~5,更优选为1:1~2。在本发明中,所述点击化学反应的温度优选为25~55℃,更优选为30~50℃,进一步优选为40~45℃,所述点击化学反应的时间优选为24~48h,更优选为25~45h,进一步优选为30~40h。在本发明中,所述点击化学反应采用的溶剂优选包括二甲基亚砜、乙腈和n,n-二甲基甲酰胺中的一种或几种,本发明对于所述溶剂的用量没有特殊限定,能够保证点击化学反应顺利进行即可。在本发明中,当所述r1基团为叠氮基团,r2基团为炔基时,所述点击化学反应优选在铜离子催化剂存在条件下进行,所述铜离子催化剂优选包括氯化亚铜、硫酸亚铜、硝酸亚铜;所述端基修饰r2基的dna与铜离子催化剂的摩尔比优选为1:1~5,更优选为1:1~2。
67.在本发明中,所述具有单链rna粘性末端的功能性核酸分子包括单链rna粘性末端和功能性核酸分子构成,rna粘性末端为功能性核酸分子中一条核酸链的延伸序列。在本发明中,所述功能性核酸分子优选包括反义寡核苷酸(aso)、小干扰核酸(sirna)或微小核糖核酸(mirna)。在本发明中,当所述功能性核酸分子为sirna或mirna时,所述单链rna粘性末
端由所述sirna或mirna中的正义链的3’端或5’端延伸引出;当所述功能性核酸分子为aso时,单链rna粘性末端由所述aso的一端延伸引出。在本发明中,所述具有单链rna粘性末端的功能性核酸分子委托生工生物工程(上海)股份有限公司合成。
68.在本发明中,所述刷状核酸组装体的制备方法优选包括以下步骤:将侧链修饰dna的可降解高分子化合物、具有单链rna粘性末端的功能性核酸分子和缓冲溶液混合,进行孵育,得到刷状核酸组装体。
69.在本发明中,所述缓冲溶液优选包括三羟甲基氨基甲烷盐酸盐缓冲液、三羟甲基氨基甲烷醋酸盐缓冲液或磷酸盐缓冲液。在本发明中,所述三羟甲基氨基甲烷盐酸盐缓冲液的ph值优选为7.5~8.5,更优选为7.8~8.2,进一步优选为8。在本发明中,所述三羟甲基氨基甲烷醋酸盐缓冲液的ph值优选为7.2~8.4,更优选为7.4~8.2,进一步优选为7.5~8。在本发明中,所述磷酸盐缓冲液的ph值优选为7.2~7.6,更优选为7.3~7.5,进一步优选为7.4。
70.在本发明中,所述侧链修饰dna的可降解高分子化合物中dna的物质的量与缓冲溶液的体积之比优选为1~1000μmol:1l,更优选为100~800μmol:1l,进一步优选为300~500μmol:1l。
71.本发明对于所述混合的方式没有特殊限定,能够将原料混合均匀即可,具体如搅拌混合。在本发明的具体实施例中,所述混合的顺序优选为将侧链修饰dna的可降解高分子化合物溶解于缓冲溶液中,得到侧链修饰dna的可降解高分子化合物溶液;将所述侧链修饰dna的可降解高分子化合物溶液与具有单链rna粘性末端的功能性核酸分子混合。
72.在本发明中,所述孵育的温度优选为室温,所述孵育的时间优选为20~60min,更优选为30~50min。在本发明中,所述孵育过程中,具有单链rna粘性末端的功能性核酸分子中的功能性核酸分子中的单链rna粘性末端与侧链修饰dna的可降解高分子化合物中的dna进行碱基互补。
73.本发明提供了一种复合纳米粒子,由上述技术方案所述的刷状核酸组装体与阳离子聚合物自组装得到。
74.在本发明中,所述阳离子聚合物包括聚β-氨基酯、聚氨基酯、聚乙烯亚胺、聚赖氨酸、聚精氨酸、聚酰胺-胺和壳聚糖中的一种或几种,更优选包括聚β-氨基酯、聚氨基酯、聚乙烯亚胺、聚赖氨酸、聚精氨酸、聚酰胺-胺或壳聚糖。
75.在本发明中,所述复合纳米粒子的制备方法优选包括以下步骤:将刷状核酸组装体、阳离子聚合物和缓冲溶液混合,进行自组装,得到复合纳米粒子。
76.在本发明中,所述缓冲溶液优选包括醋酸钠缓冲液或磷酸盐缓冲液。在本发明中,所述醋酸钠缓冲液的ph值优选为5.0~5.5,更优选为5.1~5.4,进一步优选为5.2~5.3。在本发明中,所述磷酸盐缓冲液的ph值优选为7.2~7.6,更优选为7.3~7.5,进一步优选为7.4。
77.在本发明中,所述阳离子聚合物的质量与缓冲溶液的体积之比优选为0.1~10g:1l,更优选为0.5~8g:1l,进一步优选为1~5g:1l。
78.在本发明中,所述刷状核酸组装体中核酸与阳离子聚合物的质量比优选为1:10~100,更优选为1:20~80,进一步优选为1:40~50。
79.本发明对于所述混合的方式没有特殊限定,能够将原料混合均匀即可,具体如涡
旋混合。在本发明的具体实施例中,所述混合的顺序优选为将阳离子聚合物溶解于缓冲溶液中,得到阳离子聚合物溶液;将所述阳离子聚合物溶液与刷状核酸组装体分子混合。
80.在本发明中,所述自组装的温度优选为室温,所述自组装的时间优选为10~30min,更优选为15~25min,进一步优选为20min。在本发明中,所述自组装过程中,阳离子聚合物与刷状核酸组装体之间通过静电相互作用进行自组装。
81.所述自组装后,本发明优选还包括将所述自组装得到自组装反应液进行超滤,得到复合纳米粒子。在本发明中,所述超滤用溶剂优选为磷酸盐缓冲溶液,所述磷酸盐缓冲液的ph值优选为7.2~7.6,更优选为7.3~7.5,进一步优选为7.4。
82.本发明提供了上述技术方案所述的刷状核酸组装体或上述技术方案所述的复合纳米粒子在非疾病诊断或治疗中基因调控或制备核酸药物中的应用。
83.与现有技术相比,本发明通过将难以有效压缩的寡核苷酸药物等功能核酸分子经单链rna粘性末端修饰,然后与侧链修饰dna的可降解高分子化合物经过碱基互补形成拓扑结构更大的刷状核酸组装体,增强了与阳离子聚合物相互作用,提高了功能性核酸分子的负载率,进而提高了寡核苷酸药物等功能核酸分子的递送效率。而且,刷状核酸组装体中dna/rna杂交双链部分能够被核酸酶rnase h识别和降解,从而释放出功能核酸部分,实现了高效核酸递送和基因调控效果。本发明提供的刷状核酸组装体为非病毒递送体系,免疫原性低,安全性高,成本低。
84.刷状核酸组装体与阳离子聚合物自组装形成复合纳米粒子,能够富集功能性核酸并形成高电荷密度的多价拓扑核酸结构,从而实现了在使用低剂量阳离子载体条件下高效压缩功能核酸分子并形成稳定的核酸/聚合物复合纳米粒子用于功能核酸的递送,毒性低,成本低。本发明提供的复合纳米粒子在生理条件下具有良好的稳定性,能够有效保护复合纳米粒子中所负载的功能核酸,避免被环境中核酸酶降解,能够高效地将所负载功能核酸递送至组织和细胞中。依靠复合纳米粒子中阳离子聚合物的“质子海绵”效应促进功能性核酸从内含体逃逸,从而将功能核酸高效递送至细胞质中,使得复合纳米粒子具有良好的基因调控功能。
85.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
86.实施例1
87.本实施例的聚合物2/刷状核酸组装体a复合纳米粒子的制备路线如图1所示,具体步骤如下:
88.1.1制备侧链修饰dna的可降解高分子化合物(聚合物1,dna-g-pcl)的合成
89.按照cn107281497a实施例1制备侧链修饰dna的可降解高分子化合物,其中,3端修饰二苯基环辛炔(dbco)基的dna(dbco-dna)序列为:5
’‑
ttgacctgtgaa-dbco-3’,seq id no.1。
90.聚合物1的水合粒径如图2所示,由图2可知,聚合物1的平均水合粒径为20nm。
91.1.2封端的线性聚(β-氨基酯)(聚合物2)的合成
92.1.2.1末端未修饰的聚(β-氨基酯)(中间产物3)的合成
93.将500.0mg 1,6-己二醇二丙烯酸酯和138.0mg 3-氨基-1-丙醇置于10ml烧瓶中,在90℃、无溶剂条件下混合搅拌反应12h,反应完毕后,经二氯甲烷/冰乙醚的条件下反复冲洗提纯3次,并置于真空干燥箱、50℃干燥至恒重,得到末端未修饰的聚(β-氨基酯)(中间产物3,白色粘稠液体)。
94.中间产物3的1h nmr谱图如图3所示,测试溶剂dmso-d6,各质子峰的归属如下:δ(ppm):1.25-1.35(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2),1.45-1.5(2h,t,nch2ch2ch2oh),1.5-1.65(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)),2.3-2.45(6h,m,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)and nch2ch2ch2oh),2.6-2.7(4h,t,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)),3.3-3.4(2h,m obsc,nch2ch2ch2oh),3.95-4.05(4h,t,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)),4.05-4.15(t,ch2(coo)ch=ch2),4.25-4.35(br,nch2ch2ch2oh),5.9-6(d,cooch=ch2),6.1-6.2(dd,cooch=ch2),6.3-6.4(d,cooch=ch2)。中间产物3的的凝胶渗透色谱数据图如图5所示,由图5可知,中间体3的数均分子量mn为5093,重均分子量mw为14261。
95.1.2.2聚合物2(线性聚(β-氨基酯))的合成
96.将300.0mg中间产物3溶解在1.5ml二甲基亚砜中,加入26.4mg 2-二甲氨基乙胺,在室温条件下反应12h,反应完毕后,经二氯甲烷/冰乙醚的条件下反复冲洗提纯3次,置于真空干燥箱中在50℃条件下干燥至恒重,得到封端的聚(β-氨基酯)(记为聚合物2,白色粘稠液体);将聚(β-氨基酯)溶解于二甲基亚砜中配制为100mg/ml的聚(β-氨基酯)储存溶液。
97.聚合物2的1h nmr谱图如图4所示,测试溶剂dmso-d6,各质子峰的归属如下:δ(ppm):1.25-1.35(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2),1.45-1.5(2h,t,nch2ch2ch2oh),1.5-1.65(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)),2.09-2.12(6h,s,nhch2ch2n(ch3)2),2.24-2.28(2h,t,nhch2ch2n(ch3)2),2.3-2.45(6h,m,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)and nch2ch2ch2oh),2.53-2.57(br,m,nhch2ch2n(ch3)2),2.6-2.7(4h,t,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)),2.71-2.76(2h,br,t,coch2ch2nhch2ch2n(ch3)2),3.3-3.4(2h,m obsc,nch2ch2ch2oh),3.95-4.05(4h,t,ch2ch2nch2ch2(coo)ch2ch2ch2ch2ch2ch2(coo)),4.05-4.15(t,ch2(coo)ch=ch2),4.25-4.35(br,nch2ch2ch2oh)。聚合物2的凝胶渗透色谱数据图由图5可知,聚合物2的数均分子量mn为5731,重均分子量mw为18513。
98.1.3刷状核酸组装体a(简称为siegfp-brush)的合成
99.将聚合物1溶解于ph值为7.2的三羟甲基氨基甲烷醋酸镁缓冲溶液中,得到聚合物1溶液;将带粘性末端并靶向增强型绿色荧光蛋白(egfp)的sirna(简称为siegfp,带粘性末端的正义链(tailed-sense siegfp)序列为:5
’‑
rururcrarcrargrgrurcrarargrarcrgrurarararcrgrgrcrcrarcrarargrudtdc-3’,seq id no.2;靶向egfp的反义链rna(antisense siegfp)序列为:5
’‑
rarcrururgrurgrgrcrcrgrurururarcrgrurcdgdc-3’,seq id no.3;上述序列中前缀r代表该核苷酸单元为核糖核酸单元,前缀d代表改核苷酸单元为脱氧核糖核酸单元,下划线部分为单链粘性末端部分,其序列与侧链修饰dna的可降解高分子化合物的dna序列互补)置于聚合物1溶液中混合均匀,在37℃条件下孵育15min,冷却至室温,得到刷状核酸组装体a(简称为siegfp-brush)溶液;其中,聚合物1溶液的浓度为30μm;聚合物1中dna与siegfp的摩尔比分别为1:1.3、1:1.2、1:1.1、1:1.0、1:0.9和1:0.8。
100.刷状核酸组装体a在2%的琼脂糖凝胶电泳图如图6所示,由图6可知,刷状核酸组装体a表现为单一的条带,并且条带对应泳道中未观察到游离的sirna,说明刷状核酸组装体的成功制备。
101.刷状核酸组装体a的水合粒径如图2所示,由图2可知,刷状核酸组装体a的平均水合粒径为35nm。
102.刷状核酸组装体a的透射电子显微镜如图7所示,由图7可知,刷状核酸组装体a的平均尺寸为30nm。
103.1.4聚合物2/刷状核酸组装体a复合纳米粒子的合成
104.将聚合物2溶解于醋酸钠缓冲液(ph=5.2)中,得到浓度为2mg/ml的聚合物2水溶液,利用浓度为1mol/l的盐酸将刷状核酸组装体a溶液的ph调节至5.2,将聚合物2水溶液加入到刷状核酸组装体a溶液中,涡旋10s后在室温条件下静置孵育10min,得到聚合物2/刷状核酸组装体a复合纳米粒子溶液;其中,聚合物2与带粘性末端靶向egfp的干扰小核酸(siegfp)质量比分别为10:1、20:1、30:1、40:1、50:1和100:1。
105.聚合物2/刷状核酸组装体a复合纳米粒子的水合粒径如图2所示,由图2可知,聚合物2/刷状核酸组装体a复合纳米粒子的平均水合粒径为110nm。
106.带粘性末端靶向egfp的干扰小核酸(siegfp)、刷状核酸组装体a(siegfp-brush)和聚合物2/刷状核酸组装体a复合纳米粒子在0.5%的琼脂糖凝胶中的电泳图如图8所示,由图8可知,聚合物2/刷状核酸组装体a复合纳米粒子表现为停滞在胶孔中的条带,并且随着聚合物剂量的增加,刷状核酸组装体a可以被完全压缩并停滞在胶孔里,说明聚合物2/刷状核酸组装体a复合纳米粒子成功制备。
107.聚合物2/刷状核酸组装体a复合纳米粒子的透射电子显微镜图如图10所示,由图10可知,聚合物2/刷状核酸组装体a复合纳米粒子粒径的平均尺寸在100nm。
108.对比例1
109.按照实施例1步骤1.2~1.3的方法制备,与实施例1的区别仅在于步骤1.3中将刷状核酸组装体a替换为带粘性末端靶向egfp的干扰小核酸(siegfp),得到聚合物2/siegfp复合物。
110.带粘性末端靶向egfp的干扰小核酸(siegfp)和聚合物2/siegfp复合物的0.5%琼脂糖电泳图如图9所示,由图9可知,随着聚合物2剂量的增加,聚合物2/siegfp复合物的条带几乎没有出现阻滞现象,siegfp不能被有效压缩并停滞在胶孔里,说明聚合物2与siegfp直接复合时静电相互作用弱,无法有效压缩siegfp形成结构稳定的纳米粒子。
111.测试例1
112.(1)稳定性实验
113.分别将siegfp、dna-g-pcl和聚合物2/刷状核酸组装体a复合纳米粒子与含10%fbs的dmem培养基在37℃条件下分别孵育0h、2h、4h、6h、8h、10h,加入过量的肝素钠和核糖核酸酶h,充分解离聚合物2和聚合物1,释放的siegfp采用0.5%琼脂糖凝胶电泳进行分析,结果如图11所示。由图11可知,当孵育时间增加至8h时,在琼脂糖凝胶中siegfp条带并没有明显减弱,说明聚合物2/刷状核酸组装体a复合纳米粒子能够在含10%fbs的dmem培养基中稳定存在。
114.(2)有效减缓rna酶对sirna的降解作用
115.实验组:向聚合物2/刷状核酸组装体a复合纳米粒子中加入rnasea酶,分别配置rnasea酶浓度为0.05u/ml、0.5u/ml、1u/ml、2u/ml和5u/ml的聚合物2/刷状核酸组装体a复合纳米粒子溶液,在37℃条件下孵育1h。对照组:向siegfp中加入rnase a酶配制成浓度为0.05u/ml的siegfp溶液。然后进行10%变性凝胶电泳分析,结果如图12所示。由图12可知,对照组中含有0.05u/mlrnasea酶的siegfp溶液在37℃条件下孵育5min后,siegfp被rnasea酶完全降解。rnasea酶浓度为0.5u/ml的聚合物2/刷状核酸组装体a复合纳米粒子溶液,在37℃条件下孵育1h后,siegfp只会被部分降解,说明聚合物2/刷状核酸组装体a复合纳米粒子能够有效减缓rnase酶对sirna的降解作用。
116.(3)细胞基因沉默实验
117.分别将聚合物2/刷状核酸组装体a复合纳米粒子、聚合物2/siegfp复合物和siegfp与能够持续表达增强型绿色荧光蛋白的293t细胞(293t-egfp)在37℃、5%co2条件下共培养48h后,采用流式分析术分析细胞荧光信号,结果如图13所示,由图13可知,相较于单独的sirna被阳离子聚合物压缩的样品(聚合物2/siegfp),装载有siegfp的聚合物2/刷状核酸组装体a复合纳米粒子表现出更强的基因沉默效果,且随着siegfp浓度的增加,基因沉默效率也大幅提高。
118.采用实时荧光定量pcr验证聚合物2/刷状核酸组装体a复合纳米粒子的基因沉默效率,结果如图14所示,由图14可知,聚合物2/刷状核酸组装体a复合纳米粒子实验组具有最高的基因沉默效率。
119.实施例2
120.聚合物2/刷状核酸组装体b复合纳米粒子的制备路线如图15所示,具体步骤如下:
121.2.1刷状核酸组装体b(简称为sitgfβ1-brush)的合成
122.将聚合物1溶解于ph值为7.2的三羟甲基氨基甲烷醋酸镁缓冲溶液中,得到聚合物1溶液;将带粘性末端并靶向转化生长因子tgf-β1的sirna(简称为sitgfβ1,带粘性末端的正义链(tailed-sense sitgfβ1,seq id no.4)序列为:5
’‑
rururcrarcrargrgrurcrarargrgrurgrgrarararcrcrcrarcrararcrgraradtdt-3’,反义链(antisense sitgfβ1,seq id no.5)序列为:5
’‑
rururcrgrururgrurgrgrgrurururcrcrarcrcdadt-3’;上述序列中前缀r代表该核苷酸单元为核糖核酸单元,前缀d代表改核苷酸单元为脱氧核糖核酸单元,下划线部分为单链粘性末端部分,其序列与侧链修饰dna的可降解高分子化合物的dna序列互补)置于聚合物1溶液中混合均匀,在37℃条件下孵育15min,冷却至室温,得到刷状核酸组装体b(简称为sitgfβ1-brush)溶液;其中,聚合物1溶液的浓度为30μm;聚合物1中dna与sitgfβ1的摩尔比为1:1。
123.sitgfβ1、聚合物1和刷状核酸组装体b在2%的琼脂糖凝胶电泳图如图16所示,由图16可知,刷状核酸组装体b表现为单一的条带,并且条带对应泳道中未观察到游离的sirna,说明刷状核酸组装体b的成功制备。
124.刷状核酸组装体b的水合粒径如图17所示,由图17可知,刷状核酸组装体b的平均水合粒径为40nm。刷状核酸组装体b的的透射电子显微镜图如图18所示,由图18可知,刷状核酸组装体b的平均尺寸为35nm。
125.2.2聚合物2/刷状核酸组装体b复合纳米粒子的合成
126.将聚合物2溶解于醋酸钠缓冲液(ph=5.2)中,得到浓度为2mg/ml的聚合物2水溶
液;利用浓度为1mol/l的盐酸将刷状核酸组装体b溶液ph值调节至5.2,将聚合物2水溶液加入到刷状核酸组装体b溶液中,涡旋10s后在室温条件下静置孵育10min,得到聚合物2/刷状核酸组装体b复合纳米粒子溶液;其中,聚合物2与sitgfβ1质量比分别为10:1、20:1、30:1、40:1和50:1。
127.sitgfβ1、刷状核酸组装体b和聚合物2/刷状核酸组装体b复合纳米粒子在0.5%的琼脂糖凝胶电泳图如图19所示,由图19可知,聚合物2/刷状核酸组装体b复合纳米粒子表现为停滞在胶孔中的条带,并且随着聚合物2剂量的增加,刷状核酸组装体b可以被完全压缩并停滞在胶孔里,说明聚合物2/刷状核酸组装体b复合纳米粒子成功制备。
128.聚合物2/刷状核酸组装体b复合纳米粒子的水合粒径如图17所示,由图17可知,聚合物2/刷状核酸组装体b复合纳米粒子的水合粒径为110nm。聚合物2/刷状核酸组装体b复合纳米粒子的透射电子显微镜照片如图21所示,由图21可知,聚合物2/刷状核酸组装体b复合纳米粒子的平均尺寸为100nm。
129.对比例2
130.按照实施例2制备,与实施例2的区别仅在于将刷状核酸组装体b替换为sitgfβ1,得到聚合物2/sitgfβ1复合物(pbae/sitgfβ1)。
131.sitgfβ1与聚合物2/sitgfβ1复合物的0.5%琼脂糖电泳图如图20所示,由图20可知,随着聚合物2剂量的增加,聚合物2/sitgfβ1复合物的条带几乎没有出现阻滞现象,sitgfβ1不能被有效压缩并停滞在胶孔里,说明聚合物2与sitgfβ1直接复合时静电相互作用弱,无法有效压缩sitgfβ1形成结构稳定的纳米粒子。
132.测试例2
133.(1)细胞基因沉默实验
134.分别将sitgfβ1、聚合物2/sitgfβ1复合物和聚合物2/刷状核酸组装体b复合纳米粒子与增生性瘢痕细胞在37℃、5%co2条件下共培养48h,其中,聚合物2/刷状核酸组装体b复合纳米粒子中sitgfβ1的浓度为60nm;采用实时荧光定量pcr验证sitgfβ1、聚合物2/sitgfβ1复合物和聚合物2/刷状核酸组装体b复合纳米粒子的基因沉默效率,测试结果如图22所示,由图22可知,聚合物2/刷状核酸组装体b复合纳米粒子实验组具有最高的基因沉默效率。说明,本发明制备的聚合物2/刷状核酸组装体b复合纳米粒子能够通过基因沉默抑制增生性瘢痕细胞的增殖。
135.选择转化生长因子-β1(tgfβ1)作为基因沉默的靶点,制备的聚合物2/刷状核酸组装体b复合纳米粒子、聚合物2/sitgfβ1复合物以及单独的sitgfβ1分别与增生性瘢痕细胞在37℃、5%co2条件下共培养48h后,采用pi染色方法进行细胞周期测试,结果如图23所示,由图23可知,装载有沉默tgfβ1蛋白的sirna的聚合物2/刷状核酸组装体b复合纳米粒子显示了很好的诱导细胞阻滞在g0/g1期的能力,说明本发明制备的聚合物2/刷状核酸组装体b复合纳米粒子在治疗增生性瘢痕中具有潜在的应用价值。
136.实施例3
137.聚合物3/刷状核酸组装体a复合纳米粒子的制备路线如图24所示,具体步骤如下:
138.3.1封端的支化聚(β-氨基酯)(聚合物3)的合成
139.3.1.1末端未修饰的支化聚(β-氨基酯)(中间产物4)的合成
140.将500.0mg 1,4-丁二醇二丙烯酸酯、130.0mg 5-氨基-1-戊醇和63.8mg胱胺加入
到1ml二甲基亚砜溶剂,在90℃条件下搅拌混合12h,经二氯甲烷/冰乙醚的条件下反复冲洗提纯3次,置于真空干燥箱中在25℃条件下干燥至恒重,得到末端未修饰的支化聚(β-氨基酯)(记为中间产物4,淡黄色粘稠液体)。
141.中间产物4的1h nmr谱图,如图25所示,测试溶剂dmso-d6,各质子峰的归属如下:δ(ppm):1.15-1.25(2h,br,nch2ch2ch2ch2ch2oh),1.25-1.35(2h,br,nch2ch2ch2ch2ch2oh),1.35-1.45(2h,br,nch2ch2ch2ch2ch2oh),1.55-1.7(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)),2.3-2.4(6h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)and nch2ch2ch2ch2ch2oh),2.6-2.67(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)),2.67-2.77(8h,m,nch2ch2ssch2ch2n),3.3-3.4(2h,nch2ch2ch2ch2ch2oh),3.9-4.05(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)),4.05-4.15(br,ch2(coo)ch=ch2),4.25-4.35(br,nch2ch2ch2ch2ch2oh),5.9-6(d,cooch=ch2),6.1-6.2(dd,cooch=ch2),6.3-6.4(d,cooch=ch2)。
142.3.1.2聚合物3的合成
143.将300.0mg中间产物4溶解在1.5ml二甲基亚砜中,充分搅拌溶解后,加入47.2mg1-(3-氨丙基)-4-甲基哌嗪,在室温下条件下反应12h,经二氯甲烷/冰乙醚的条件下反复冲洗提纯3次,置于真空干燥箱中在在25℃条件下干燥至恒重,得到封端的支化聚(β-氨基酯)(记为聚合物3,白色粘稠液体)。将聚合物3溶解于二甲基亚砜中配制为100mg/ml的聚合物3储存溶液备用。
144.聚合物3的1h nmr谱图,如图26所示,测试溶剂dmso-d6,各质子峰的归属如下:δ(ppm):1.15-1.25(2h,br,nch2ch2ch2ch2ch2oh),1.25-1.35(2h,br,nch2ch2ch2ch2ch2oh),1.35-1.45(2h,br,nch2ch2ch2ch2ch2oh),1.50(2h,quint,-nhch2ch2ch2n《(ch2ch2》nch3)),1.55-1.7(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)),2.13(3h,s,-nhch2ch2ch2n《(ch2ch2》nch3),2.3-2.4(6h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)and nch2ch2ch2ch2ch2oh and-nhch2ch2ch2n《(ch2ch2》nch
3 and-nhch2ch2ch2n《(ch2ch2》nch3),2.47(2h,t,-nhch2ch2ch2n《(ch2ch2》nch3)2.6-2.67(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)),2.67-2.77(8h,m,nch2ch2ssch2ch2n),3.3-3.4(2h,nch2ch2ch2ch2ch2oh),3.9-4.05(4h,br,ch2ch2nch2ch2(coo)ch2ch2ch2ch2(coo)),4.25-4.35(br,nch2ch2ch2ch2ch2oh)。聚合物3的凝胶渗透色谱数据图如图27所示,由图27可知,聚合物3的数均分子量mn为2493,重均分子量mw为8846。
145.3.2聚合物3/刷状核酸组装体a复合纳米粒子的合成
146.按照实施例1步骤1.3~1.4制备,与实施例1的区别在于,步骤1.4中将聚合物2替换为聚合物3,聚合物3与刷状核酸组装体a中siegfp质量比分别为10:1、20:1、30:1、40:1和50:1。
147.siegfp、siegfp-brush和聚合物3/刷状核酸组装体a复合纳米粒子在0.5%的琼脂糖凝胶电泳图如图28所示,由图28可知,聚合物3/刷状核酸组装体a复合纳米粒子表现为停滞在胶孔中的条带,并且随着聚合物3剂量的增加,刷状核酸组装体a可以被完全压缩并停滞在胶孔里,说明聚合物3/刷状核酸组装体a复合纳米粒子成功制备。
148.聚合物3/刷状核酸组装体a复合纳米粒子的透射电子显微镜图如图29所示,由图29可知,聚合物3/刷状核酸组装体a复合纳米粒子粒径的平均尺寸约为100nm。
149.对比例3
150.按照实施例3法制备,与实施例3的区别仅在于步骤1.2中将刷状核酸组装体a替换为siegfp,得到聚合物3/siegfp复合物。
151.测试例3
152.细胞基因沉默实验
153.分别将siegfp、聚合物3/刷状核酸组装体a复合纳米粒子和聚合物3/siegfp复合物与293t-egfp细胞在37℃、5%co2条件下共培养48h,其中,siegfp、聚合物3/刷状核酸组装体a复合纳米粒子和聚合物3/siegfp复合物中sirna浓度均为60nm(60nmol/l),采用流式分析术分析细胞荧光信号,结果如图30所示,由图30可知,在相同的sirna浓度(60nm)下,聚合物3/siegfp复合物几乎没有基因沉默效果,装载有siegfp的聚合物3/刷状核酸组装体b复合纳米粒子表现出更强的基因沉默效果。
154.实施例4
155.聚合物4/刷状核酸组装体a复合纳米粒子的制备路线如图31所示,具体步骤如下:
156.按照实施例1步骤1.3~1.4制备,与实施例1的区别在于,步骤1.4中将聚合物2替换为聚合物4(聚乙烯亚胺,pei),聚合物4与刷状核酸组装体a中siegfp质量比分别为1:1、1.5:1、3:1、5:1和10:1,得到聚合物4/刷状核酸组装体a复合纳米粒子溶液。
157.聚合物4/刷状核酸组装体a复合纳米粒子在0.5%的琼脂糖凝胶电泳图如图32所示,由图32可知,聚合物4/刷状核酸组装体a复合纳米粒子表现为停滞在胶孔中的条带,并且随着聚合物4剂量的增加,刷状核酸组装体a可以被完全压缩并停滞在胶孔里,说明聚合物4/刷状核酸组装体a复合纳米粒子成功制备。
158.聚合物4/刷状核酸组装体a复合纳米粒子的水合粒径如图33所示,由图33可知,聚合物4/刷状核酸组装体a复合纳米粒子的水合粒径为100nm。聚合物4/刷状核酸组装体a复合纳米粒子的透射电子显微镜图如图34所示,由图34可知,聚合物4/刷状核酸组装体a复合纳米粒子的平均尺寸为90nm。
159.对比例4
160.按照实施例4法制备,与实施例4的区别仅在于将刷状核酸组装体a替换为siegfp,得到聚合物4/siegfp复合物。
161.测试例4
162.细胞基因沉默实验
163.分别将siegfp、聚合物4/刷状核酸组装体a复合纳米粒子和聚合物4/siegfp复合物与293t-egfp在37℃、5%co2条件下细胞共培养48h,其中,siegfp、聚合物4/刷状核酸组装体a复合纳米粒子和聚合物4/siegfp复合物的浓度均为60nmol/l,采用流式分析术分析细胞荧光信号,结果如图35所示,由图35可知,在相同的sirna浓度下,相较于单独的sirna被pei聚合物压缩的样品(聚合物4/siegfp复合物)来说,装载有siegfp的聚合物4/刷状核酸组装体a复合纳米粒子表现出更强的基因沉默效果。
164.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1