一种大戟科二萜PepluanolA的合成方法
一种大戟科二萜pepluanol a的合成方法
技术领域
1.本发明属于有机合成领域,具体涉及一种大戟科二萜pepluanol a的合成方法。
背景技术:
2.天然产物作为大自然的宝贵财富,在人类社会的发展中发挥了重要的作用。基于天然产物的药物开发一直是药物开发的重中之重。有统计显示,fda历年批准的临床新药中,超过一半来自天然产物或其衍生物。我国拥有丰富的天然产物资源以及悠久的中医药理论,两者的结合对于研发具有自主知识产权的新药具有重大意义。
3.来自大戟科的药用植物,在传统中医里被用于治疗哮喘和牛皮癣等疾病,其中发挥重要活性作用的往往是大戟科天然产物。由于大戟科植物天然产物结构以及生物活性的多样性,往往是发现天然药物的主要来源途径。但通过分离得到的天然产物量通常很少,无法大批量制备,因此,天然产物全合成就成为衔接活性成分发现和药物化学研究的重要中间环节。
4.二萜类化合物是一类结构复杂多样并具有重要生物活性的天然产物,设计并发展新策略、新方法,实现其简便高效合成,对于推动有机合成新方法、新理论的发展以及新药发现具有重要科学和实际意义,现有技术中报道了部分二萜类化合物的合成方法,如:公开号为cn108503652a的中国专利文献公开了一种巴豆烷二萜prostratin的化学全合成制备方法,该发明通过易制备的5-(羟甲基)-2-环戊烯-1-醇为原料,经过光照氧化去芳化反应,分子内诱导加成反应,烯烃复分解反应等关键反应,最后经过官能团转化得到目标产物。
5.公开号为cn105152992a的中国专利文献公开了一种cyclopiane四环二萜天然产物(conidiogenoneb,conidiogenone和conidiogenol)的合成方法,由已知的共轭酯出发,先合成具有共轭烯酮和环丁醇结构的中间体,再利用酸作用下重排反应,构建出四环二萜的三环骨架结构,之后经过结构修饰和酸作用下环化反应,构建出四环二萜的四环骨架结构,再以此四环二萜类骨架为关键中间体,经多步化学转化合成cyclopiane四环二萜天然产物。
6.大戟科二萜pepluanol a最初由euphorbia peplus的丙酮提取物分离得到,该化合物具有[5,4,7,3]四环骨架,共含7个手性中心,其中6个连续手性中心以及一个季碳,形成高度紧凑的稠环骨架,合成较为困难,且该化合物表现出阻断kv1.3钾离子通道的生物活性,可以潜在治疗一些t细胞介导的免疫疾病,如i型糖尿病以及哮喘等。
技术实现要素:
[0007]
本发明提供了一种大戟科二萜pepluanol a的合成方法,从外消旋原料出发,各步反应位点可控,总收率较高,达到2.5%。实现了大戟科二萜pepluanol a的17步全合成,为进一步研究大戟科二萜类化合物的构效关系打下基础。
[0008]
具体采用的技术方案如下:
[0009]
一种大戟科二萜pepluanol a的合成方法,包括以下步骤:
[0010]
步骤1:将烯酮化合物2溶解于有机溶剂中,加入叠氮三甲基硅烷、碘代试剂和有机碱,反应得到烯基碘类化合物3;
[0011]
步骤2:将烯基碘类化合物3溶解于有机溶剂中,再加入有机膦、金属钯催化剂和有机锌试剂,反应得到烯酮类化合物4;
[0012]
步骤3:将烯酮类化合物4与共轭二烯醚化合物混合,反应得到混合物并用有机溶剂溶解,再加入盐酸,进一步反应得到二酮类化合物6;
[0013]
步骤4:将二酮类化合物6溶解于有机溶剂中,加入三氟甲磺酰基化试剂和强碱,反应得到三氟甲磺酸酯类化合物7;
[0014]
步骤5:将三氟甲磺酸酯类化合物7溶解于有机溶剂中,加入金属铜催化剂和甲基金属试剂,反应得到共轭二烯类化合物8;
[0015]
步骤6:将共轭二烯类化合物8溶解于有机溶剂中,加入无机碱,反应得到半缩酮类化合物9;
[0016]
步骤7:将半缩酮类化合物9溶解于有机溶剂中,加入水、无机碱和过氧化试剂,反应得到环氧类化合物10;
[0017]
步骤8:将环氧类化合物10溶解于有机溶剂中,加入有机膦和偶氮二羧酸酯类化合物,反应得到三烯类化合物11;
[0018]
步骤9:将三烯类化合物11溶解于有机溶剂中,加入水、有机碱、催化量氧化剂和当量氧化剂,反应得到醛类化合物12;
[0019]
步骤10:将有机金属钛试剂与还原金属混合后,再加入有机碱盐酸盐和醛类化合物12,反应得到二醇类化合物13;
[0020]
步骤11:将二醇类化合物13溶解于有机卤代溶剂中,加入相转移催化剂和无机强碱,反应得到环丙烷类化合物14;
[0021]
步骤12:将环丙烷类化合物14、碘甲烷、甲基金属试剂和有机碱反应得到偕二甲基环丙烷类化合物15;
[0022]
步骤13:将偕二甲基环丙烷类化合物15溶解于混合溶剂中,再加入氮氧化物、相转移催化剂、无机碱和氧化剂,反应得到偕二甲基二酮类化合物16;
[0023]
步骤14:将偕二甲基二酮类化合物16溶解于有机溶剂中,加入有机碱和硅基化试剂,反应得到偕二甲基硅醚类化合物17;
[0024]
步骤15:将偕二甲基硅醚类化合物17溶解于有机溶剂中,加入金属钯催化剂,反应得到偕二甲基烯酮类化合物18;
[0025]
步骤16:将偕二甲基烯酮类化合物18溶解于有机溶剂中,加入甲基金属试剂,反应得到偕二甲基烯丙醇类化合物19;
[0026]
步骤17:将偕二甲基烯丙醇类化合物19溶解于有机溶剂中,再加入硅胶、无机盐和氧化剂,搅拌后,加入盐酸后继续搅拌,得到式i所示结构的大戟科二萜pepluanol a;
[0027][0028]
优选的,步骤1中,所述的碘代试剂为单质碘或n-碘代丁二酰亚胺;所述的有机碱为吡啶、三乙胺、4-二甲氨基吡啶或1,4-二氮杂二环[2.2.2]辛烷;烯酮化合物2、有机碱、碘代试剂和叠氮三甲基硅烷的摩尔比为1:4~5:1~1.5:1~1.5;烯酮化合物2与有机溶剂的配比为1mmol:1~5ml。
[0029]
优选的,步骤2中,所述的有机膦为三(2-呋喃基)膦、三苯基膦或三丁基膦;所述的金属钯催化剂为双二亚苄基丙酮钯、醋酸钯、四(三苯基膦)钯、氯化钯或双(乙腈)二氯化钯;所述的有机锌试剂为3-丁烯基溴化锌;烯基碘类化合物3与有机溶剂的配比为1mmol:3~5ml;烯基碘类化合物3、有机锌试剂、有机膦和金属钯催化剂的摩尔比为1:1.0~1.5:0.1~0.5:0.05~0.1。
[0030]
优选的,步骤3中,所述的共轭二烯醚化合物为rawal双烯;烯酮类化合物4和共轭二烯醚化合物的摩尔比为1:1.5~2。
[0031]
优选的,步骤4中,所述的强碱为双(三甲基硅烷基)氨基钾、双(三甲基硅烷基)氨基锂、双(三甲基硅烷基)氨基钠或二异丙基氨基锂;所述的三氟甲磺酰基化试剂为n-苯基双(三氟甲烷磺酰)亚胺、2-[n,正双(三氟甲烷烷磺酰)氨基]-5-氯吡啶或三氟甲磺酸酐;二酮类化合物6与有机溶剂的配比为1mmol:5~10ml;二酮类化合物6、强碱和三氟甲磺酰基化试剂的摩尔比为1:1.1~1.5:1.1~1.5。
[0032]
优选的,步骤5中,所述的金属铜催化剂为碘化亚铜、溴化亚铜或溴化亚铜二甲硫醚;所述的甲基金属试剂为甲基溴化镁、三甲基铝、甲基碘化镁或甲基锂;三氟甲磺酸酯类化合物7与有机溶剂的配比为1mmol:5~10ml;三氟甲磺酸酯类化合物7、金属铜催化剂和甲基金属试剂的摩尔比为1:0.05~0.1:1.5~2。
[0033]
优选的,步骤6中,所述的无机碱为乙醇钠、碳酸钾、甲醇钠、叔丁醇钾或叔丁醇钠;共轭二烯类化合物8与无机碱的摩尔比为1:3;所述共轭二烯类化合物8与有机溶剂的配比为1mmol:5~10ml。
[0034]
优选的,步骤7中,所述的无机碱为碳酸氢钠、碳酸氢钾或乙酸钠;所述的过氧化试剂为过氧单磺酸钾、双氧水、间氯过氧苯甲酸或过氧丙酮;半缩酮类化合物9与有机溶剂的配比为1mmol:5~10ml;半缩酮类化合物9、无机碱和过氧化试剂的摩尔比为1:5~10:1~5。
[0035]
优选的,步骤8中,所述的有机膦为三(2-呋喃基)膦、三苯基膦或三丁基膦;所述的偶氮二羧酸酯类化合物为偶氮二甲酸二乙酯或偶氮二甲酸二异丙酯;环氧类化合物10与有机溶剂的配比为1mmol:10~15ml;环氧类化合物10、有机膦和偶氮二羧酸类化合物的摩尔比为1:2~4:2~4。
[0036]
优选的,步骤9中,所述的有机碱为2,6-二甲基吡啶、吡啶或四甲基乙二胺;所述的催化量氧化剂为四氧化锇或锇酸钾;所述的当量氧化剂为高碘酸或高碘酸钠;三烯类化合
物11与有机溶剂的配比为1mmol:12~17ml;三烯类化合物11、有机碱、催化量氧化剂和当量氧化剂的摩尔比为1:1~3:0.05~0.15:3~5。
[0037]
优选的,步骤10中,所述的有机金属钛试剂为二氯二茂钛;所述的还原金属为锌粉或锰粉;所述的有机碱盐酸盐为2,4,6-三甲基吡啶盐酸盐;醛类化合物12、还原金属、有机金属钛试剂和有机碱盐酸盐的摩尔比为1:1~5:1:1。
[0038]
优选的,步骤11中,所述的相转移催化剂为苄基三乙基氯化铵、四丁基氯化铵、四丁基溴化铵或四丁基碘化铵;所述的无机强碱为叔丁醇钾、氢氧化钠或氢氧化钾;二醇类化合物13与有机卤代溶剂的配比为1mmol:20~50ml;二醇类化合物13、相转移催化剂和无机强碱的质量比为1:0.05~0.1:50。
[0039]
优选的,步骤12中,所述的甲基金属试剂为甲基锂或甲基铜锂;所述的有机碱为六甲基磷酰三胺或n,n-二甲基丙烯基脲;环丙烷类化合物14、甲基金属试剂、碘甲烷和有机碱的摩尔比为1:30~50:30~50:5~20。
[0040]
优选的,步骤13中,所述的无机碱为碳酸氢钠、碳酸氢钾或乙酸钠;所述的相转移催化剂为苄基三乙基氯化铵、四丁基氯化铵、四丁基溴化铵或四丁基碘化铵;所述的氧化剂为氯代丁二酰亚胺、次氯酸钠或醋酸碘苯;所述的氮氧化物为四甲基哌啶氮氧化物或2-氮杂金刚烷-n-氧自由基;偕二甲基环丙烷类化合物15与混合溶剂的配比为1mmol:100~150ml;偕二甲基环丙烷类化合物15、无机碱、相转移催化剂、氧化剂和氮氧化物的物质的量比为1:4~6:1:1~3:0.05~0.2。
[0041]
优选的,步骤14中,所述的有机碱为三乙胺、2,6-二甲基吡啶或吡啶;所述的硅基化试剂为三氟甲磺酸三甲基硅酯、三氟甲磺酸三乙基硅酯或叔丁基二甲硅基三氟甲磺酸酯;偕二甲基二酮类化合物16与有机溶剂的配比为1mmol:50~100ml;偕二甲基二酮类化合物16、有机碱和硅基化试剂的摩尔比为1:5~10:2~5。
[0042]
优选的,步骤15中,所述的金属钯催化剂为醋酸钯或三氟醋酸钯;偕二甲基硅醚类化合物17与有机溶剂的配比为1mmol:50~100ml;偕二甲基硅醚类化合物17和金属钯催化剂的摩尔比为1:1.5~3。
[0043]
优选的,步骤16中,所述的甲基金属试剂为甲基溴化镁、甲基碘化镁、三甲基铝或甲基锂;偕二甲基烯酮类化合物18与有机溶剂的配比为1mmol:50~100ml;偕二甲基烯酮类化合物18与甲基金属试剂的摩尔比为1:1~5。
[0044]
优选的,步骤17中,所述的无机盐为乙酸钠或碳酸氢钠;所述的氧化剂为氯铬酸吡啶鎓盐或重铬酸吡啶鎓;偕二甲基烯丙醇类化合物19与有机溶剂的配比为1mmol:50~100ml;偕二甲基烯丙醇类化合物19、硅胶、无机盐和氧化剂的质量比为1:1~1.5:0.5~1:0.5~1。
[0045]
进一步优选的,步骤10中,所述的还原金属为锰粉;步骤12中,所述的有机碱为n,n-二甲基丙烯基脲;锰粉和n,n-二甲基丙烯基脲的选用都可以在保证产率的同时提高操作安全性;步骤15中,所述的金属钯催化剂为三氟醋酸钯,该条件的选择可以使得产率到达95%,三氟的强吸电子特性可以使得三氟醋酸根负离子有更强的碱性,使得钯的β氢消除更加高效。化学选择性和立体选择性一直是精细有机合成发展的关键因素,反应的位点选择性与产率和催化剂、配体、底物自身的性质等具有密不可分的联系。
[0046]
与现有技术相比,本发明的有益效果在于:
[0047]
(1)本发明所采用的合成路线是以易制备的环庚烯酮衍生物为起始原料,经过negishi偶联反应,diels-alder反应,钛自由基催化的串联环化反应等关键步骤构建关键三环骨架,最后通过后期官能团转化得到目标产物大戟科二萜pepluanol a。
[0048]
(2)本发明合成方法操作简便,17步合成得到产物,条件温和,总产率达到2.5%。合成的产物与天然产物的nmr谱图数据一致。
[0049]
(3)本发明的合成路线设计思路新颖,原料廉价易得,各个重要官能团兼容性强,方便合成多种含相同5/6/7稠环骨架的大戟科二萜结构的衍生物,为该类化合物的构效关系研究打下基础。
附图说明
[0050]
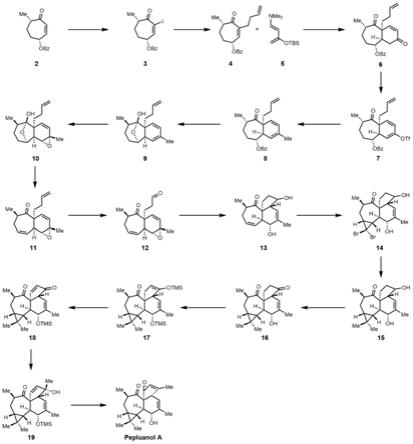
图1为本发明中大戟科二萜pepluanol a化合物的合成路线图。
[0051]
图2为本发明中大戟科二萜pepluanol a化合物的氢谱。
[0052]
图3为本发明中大戟科二萜pepluanol a化合物的碳谱。
具体实施方式
[0053]
下面结合附图与实施例,进一步阐明本发明。应理解,这些实施例仅用于说明本发明,而不用于限制本发明的范围。
[0054]
实施例1
[0055]
pepluanol a化合物的合成路线图如图1所示。
[0056]
步骤1:
[0057][0058]
在0℃条件下,向(20g,0.082mol)的烯酮化合物2的二氯甲烷(200ml)溶液中加入叠氮基三甲基硅烷(11.9ml,0.09mol),搅拌2小时后,依次加入碘单质(22.86g,0.09mol)和吡啶(26.5ml,0.33mol),混合液缓慢升至室温并搅拌24小时,50ml饱和氯化铵溶液淬灭反应,经二氯甲烷萃取,饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为烯基碘类化合物3(26.7g,88%)。
[0059]
核磁数据:
[0060]1h nmr(400mhz,cdcl3)δ=8.06(dd,j=8.2,1.1hz,2h),7.65
–
7.56(m,1h),7.53(dd,j=2.8,1.1hz,1h),7.50
–
7.44(m,2h),5.78(dt,j=10.2,2.9hz,1h),2.94
–
2.84(m,1h),2.28
–
2.21(m,1h),1.99(ddddd,j=20.2,16.9,13.3,9.9,6.7hz,2h),1.64
–
1.52(m,1h),1.23ppm(d,j=6.5hz,3h)。
[0061]
13
c nmr(100mhz,cdcl3)δ=198.8,165.4,153.8,133.5,129.8(2c),129.4,128.6(2c),105.6,73.3,43.8,31.3,27.0,17.3ppm。
[0062]
高分辨质谱数据:
[0063]
hrms(esi):calcd for c
15h16
io
3+
[m+h]
+
371.0139,found 371.0137。
[0064]
步骤2:
[0065][0066]
–
78℃条件下,向无水氯化锌(8.08g,59.4mmol)的四氢呋喃(100ml)溶液中缓慢加入3-丁烯基溴化镁(0.5m四氢呋喃溶液,118.8ml,59.4mmol),混合液缓慢升至0℃,得到3-丁烯基溴化锌试剂备用。
[0067]
向烯基碘类化合物3(20g,54.0mmol)的n,n-二甲基甲酰胺(200ml)溶液中依次加入三(2-呋喃基)膦(3.1g,5.4mmol)和双二亚苄基丙酮钯(626.4mg,2.7mmol),混合液搅拌10min,随后缓慢滴加上述3-丁烯基溴化锌试剂(220ml,0.054mol),搅拌1小时后,50ml饱和氯化铵淬灭反应,乙醚萃取,饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为烯酮类化合物4(11.9g,74%)。
[0068]
核磁数据:
[0069]1h nmr(400mhz,cdcl3)δ=8.10
–
8.03(m,2h),7.61
–
7.53(m,1h),7.50
–
7.39(m,2h),6.38(dd,j=2.5,1.2hz,1h),5.84
–
5.67(m,2h),5.02
–
4.90(m,2h),2.79
–
2.65(m,1h),2.50(ddt,j=15.1,8.2,1.2hz,1h),2.29(ddt,j=12.3,11.3,5.8hz,1h),2.25
–
2.11(m,3h),2.00
–
1.77(m,2h),1.54
–
1.42(m,1h),1.14ppm(d,j=6.5hz,3h)。
[0070]
13
c nmr(100mhz,cdcl3)δ=205.1,165.7,143.1,140.2,137.7,133.2,130.0,129.7(2c),128.4(2c),115.3,72.2,45.5,33.1,32.8,31.8,27.1,16.5ppm。
[0071]
高分辨质谱数据:
[0072]
hrms(esi):calcd for c
19h23o3+
[m+h]
+
299.1642,found 299.1642。
[0073]
步骤3:
[0074][0075]
向上式所示的烯酮类化合物4(10g,34mmol)加入rawal双烯5(13.3ml,51mmol),反应液在40℃下搅拌24小时,所得混合物溶于四氢呋喃(200ml),冷却至
–
30℃,接着缓慢滴加盐酸(2m水溶液,68ml,136mol),混合液缓慢升至室温并搅拌24小时,100ml饱和碳酸氢钠淬灭反应,乙酸乙酯萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到白色粉末状固体,即为二酮类化合物6(10.3g,83%)。
[0076]
核磁数据:
[0077]1h nmr(400mhz,cdcl3)δ=7.96(dd,j=5.1,3.3hz,2h),7.61
–
7.53(m,1h),7.43(dd,j=10.7,4.7hz,2h),6.80(d,j=10.3hz,1h),6.21(d,j=10.3hz,1h),5.73
–
5.60(m,1h),5.26
–
5.17(m,1h),4.97
–
4.85(m,2h),2.99(dp,j=8.7,6.5hz,1h),2.73(dt,j=5.8,
4.2hz,1h),2.69
–
2.57(m,2h),2.31(dt,j=10.3,8.0hz,1h),2.04
–
1.90(m,4h),1.90
–
1.73(m,2h),1.60(ddd,j=14.5,9.1,4.6hz,1h),1.15ppm(d,j=6.6hz,3h)。
[0078]
13
c nmr(100mhz,cdcl3)δ=211.9,196.5,165.4,150.7,137.7,133.3,123.0,129.7,129.7(2c),128.5(2c),115.3,75.2,55.5,44.5,43.3,38.6,37.2,29.1,29.0,27.6,17.3ppm。
[0079]
高分辨质谱数据:
[0080]
hrms(esi):calcd for c
23h27o4+
[m+h]
+
367.1904,found 367.1905。
[0081]
步骤4:
[0082][0083]
向上式所示的二酮类化合物6(11.94g,33.9mmol)的四氢呋喃(200ml)溶液中加入n-苯基双(三氟甲烷磺酰)亚胺(13.33g,37.3mmol),混合液冷却至
–
78℃,缓慢滴加双(三甲基硅烷基)氨基钾(1m四氢呋喃溶液,37.3ml,37.3mmol),缓慢升至室温并搅拌1小时,50ml饱和氯化铵淬灭反应,乙酸乙酯萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为三氟甲磺酸酯类化合物7(15.6g,29.5mmol)。
[0084]
步骤5:
[0085][0086]
向上式所示的三氟甲磺酸酯类化合物7(15.6g,29.5mmol)的四氢呋喃(200ml)溶液中加入碘化亚铜(280mg,1.48mmol),混合液冷却至0℃,缓慢滴加甲基溴化镁(3m乙醚溶液,19.7ml,59mmol),反应液搅拌3小时,50ml饱和氯化铵淬灭反应,乙醚萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为共轭二烯类化合物8(8.9g,步骤4和步骤5的产率总计为72%)。
[0087]
核磁数据:
[0088]1h nmr(400mhz,cdcl3)δ=8.01
–
7.92(m,2h),7.54(t,j=7.4hz,1h),7.45(t,j=7.5hz,2h),5.91(d,j=10.3hz,1h),5.77
–
5.61(m,1h),5.44(t,j=5.7hz,1h),5.23(d,j=9.6hz,2h),4.86(ddd,j=13.7,11.4,1.4hz,2h),3.13
–
3.01(m,2h),2.11
–
1.88(m,5h),1.82(s,3h),1.63
–
1.47(m,2h),1.45
–
1.34(m,1h),1.08ppm(d,j=6.6hz,3h)。
[0089]
13
c nmr(100mhz,cdcl3)δ=212.5,165.6,138.8,133.1,129.9,129.8(2c),129.6,128.5(2c),128.1,127.9,120.2,114.2,75.5,54.2,43.9,42.0,37.7,29.3,28.1,27.9,
21.6,17.8ppm。
[0090]
高分辨质谱数据:
[0091]
hrms(esi):calcd for c
24h29o3+
[m+h]
+
365.2111,found 365.2114。
[0092]
步骤6:
[0093][0094]
向上式所示的共轭二烯类化合物8(10g,27.5mmol)的甲醇(200ml)溶液中分批加入甲醇钠(4.5g,82.5mmol),反应液回流搅拌8小时,50ml饱和氯化铵淬灭反应,乙醚萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为半缩酮类化合物9(5.9g,22.8mmol)。
[0095]
步骤7:
[0096][0097]
向上式所示的半缩酮类化合物9(5.9g,22.8mmol)溶于丙酮(120ml)溶液中,依次加入水(30ml),碳酸氢钠(9.6g,114mmol)和过氧单磺酸钾(21g,68.4mmol),反应液搅拌1小时后,100ml饱和碳酸氢钠淬灭反应,乙醚萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为环氧类化合物10(4.4g,步骤6和步骤7的总产率为58%)。
[0098]
核磁数据:
[0099]1h nmr(400mhz,cdcl3)δ=5.84(d,j=10.3hz,1h),5.82
–
5.72(m,1h),5.56(d,j=10.4hz,1h),5.03
–
4.95(m,1h),4.94
–
4.88(m,1h),4.44(dd,j=8.3,2.5hz,1h),2.98(s,1h),2.86(d,j=8.3hz,1h),2.82(s,1h),1.97(ddd,j=11.9,8.9,2.5hz,2h),1.87
–
1.74(m,2h),1.69
–
1.56(m,5h),1.45(s,3h),1.00ppm(d,j=7.3hz,3h)。
[0100]
13
c nmr(100mhz,cdcl3)δ=139.0,133.2,126.3,114.3,106.0,74.4,57.6,52.7,48.4,43.7,42.7,36.4,28.1,27.5,25.5,21.1,15.8ppm。
[0101]
高分辨质谱数据:
[0102]
hrms(esi):calcd for c
17h25o3+
[m+h]
+
277.1798,found 277.1796。
[0103]
步骤8:
[0104][0105]
向上式所示的环氧类化合物10(1g,3.6mmol)的四氢呋喃(50ml)溶液中依次加入三苯基膦(3.8g,14.4mmol)和偶氮二甲酸二乙酯(2.2ml,14.4mmol),反应液在60℃搅拌1小时,25ml饱和碳酸氢钠淬灭反应,乙醚萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为三烯类化合物11(622mg,67%)。
[0106]
核磁数据:
[0107]1h nmr(400mhz,cdcl3)δ=6.44(d,j=10.4hz,1h),5.91(d,j=10.4hz,1h),5.67(ddt,j=16.9,10.2,6.6hz,1h),5.57
–
5.47(m,1h),5.21(dddd,j=11.6,4.5,2.6,1.7hz,1h),4.98
–
4.81(m,2h),3.66(dp,j=12.6,6.3hz,1h),3.56(s,1h),3.41(d,j=3.0hz,1h),2.47(ddq,j=18.1,5.8,2.8hz,1h),2.35
–
2.20(m,1h),2.08
–
1.86(m,2h),1.67(ddd,j=16.6,10.3,4.4hz,2h),1.52(s,3h),1.10(d,j=6.4hz,3h)。
[0108]
13
c nmr(100mhz,cdcl3)δ=211.6,138.0,131.8,130.6,127.7,127.6,114.6,66.0,59.2,52.6,40.9,38.1,37.7,36.4,29.2,21.4,16.8ppm。
[0109]
高分辨质谱数据:
[0110]
hrms(esi):calcd for c
17h23o2+
[m+h]
+
259.1693,found 259.1697。
[0111]
步骤9:
[0112][0113]
向上式所示的三烯类化合物11(500mg,1.94mmol)的1,4-二氧六环(30ml)溶液中依次加入水(10ml),2,6-二甲基吡啶(450μl,3.88mmol),四氧化锇(2%质量分数的水溶液,2.54ml,0.2mmol)和高碘酸钠(1.67g,7.76mmol),混合液搅拌3小时,25ml饱和硫代硫酸钠淬灭反应,二氯甲烷萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为醛类化合物12(353mg,70%)。
[0114]
核磁数据:
[0115]1h nmr(400mhz,cdcl3)δ=9.63(t,j=1.2hz,1h),6.36(dd,j=10.4,0.8hz,1h),5.96(d,j=10.4hz,1h),5.59
–
5.48(m,1h),5.26
–
5.16(m,1h),3.69(dt,j=18.4,6.2hz,1h),3.62(s,1h),3.44(d,j=3.1hz,1h),2.55
–
2.43(m,2h),2.42
–
2.30(m,1h),2.19
–
2.07(m,1h),2.01
–
1.89(m,2h),1.52(s,3h),1.08(d,j=6.4hz,3h)。
[0116]
13
c nmr(100mhz,cdcl3)δ=211.2,201.2,130.8,130.6,129.0,127.4,65.91,58.3,52.5,39.3,38.2,37.6,36.1,32.6,21.3,16.8ppm。
[0117]
高分辨质谱数据:
[0118]
hrms(esi):calcd for c
16h21o3+
[m+h]
+
261.1485,found 261.1482。
[0119]
步骤10:
[0120][0121]
将二氯二茂钛(191mg,0.77mmol)和锰粉(170mg,3.08mmol)溶于四氢呋喃(25ml)溶液中,剧烈搅拌10分钟后,溶液变绿,加入2,4,6-三甲基吡啶盐酸盐(151mg,0.77mmol),搅拌5分钟,接着在1小时内缓慢滴加上式所示的醛类化合物12(200mg,0.77mmol)的四氢呋喃(15ml)溶液,反应液搅拌2小时,硅藻土过滤,滤液减压浓缩,用硅胶柱层析法分离提纯产物得到白色粉末状固体,即为二醇类化合物13(199mg,99%,非对映异构体比例为1:1)。
[0122]
核磁数据:
[0123]1h nmr(400mhz,cdcl3)δ=5.60(d,j=5.4hz,2h),5.53
–
5.43(m,2h),5.18
–
4.98(m,2h),4.31(s,1h),4.22
–
4.13(m,1h),4.05(d,j=7.1hz,1h),3.93(dd,j=13.2,3.6hz,2h),3.82
–
3.70(m,2h),3.61(dt,j=12.1,6.0hz,1h),3.53(t,j=5.1hz,2h),3.45(d,j=2.0hz,1h),2.85(s,1h),2.69
–
2.55(m,2h),2.51
–
2.35(m,2h),2.15(s,1h),2.11
–
1.94(m,3h),1.94
–
1.90(m,3h),1.85(d,j=4.6hz,1h),1.84
–
1.81(m,4h),1.81
–
1.70(m,3h),1.59
–
1.46(m,2h),1.34
–
1.26(m,1h),1.18
–
1.09(m,6h)。
[0124]
13
c nmr(100mhz,cdcl3)δ=218.6,212.0,139.4,131.7,131.3,131.1,129.2,128.8,127.5,123.2,79.4,75.0,74.1,73.2,62.2,62.2,48.5,43.8,42.6,42.5,39.6,39.1,37.4,37.2,34.3,33.7,33.2,32.0,22.6,21.5,17.5,17.4ppm。
[0125]
高分辨质谱数据:
[0126]
hrms(esi):calcd for c
16h23o3+
[m+h]
+
263.1642,found 263.1644。
[0127]
步骤11:
[0128][0129]
向上式所示的二醇类化合物13(50mg,0.19mmol,非对映异构体比例为1:1)的三溴甲烷(5ml)溶液中依次加入苄基三乙基氯化铵(4.3mg)和氢氧化钠(50%质量分数的水溶液,5ml),反应液在50℃搅拌2小时,在0℃下用10ml饱和氯化铵溶液淬灭反应,二氯甲烷萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为环丙烷类化合物14。
[0130]
步骤12:
[0131][0132]
将新干燥的硫氰酸亚铜(413mg,3.4mmol)的乙醚(30ml)溶液冷却至
–
78℃,接着缓慢滴加甲基锂(1.6m乙醚溶液,4.3ml,6.8mmol),反应液缓慢升至
–
20℃,向反应液中缓慢滴加如上式所示的环丙烷类化合物14(71mg,0.17mmol)和n,n-二甲基丙烯基脲(217μl,1.7mmol)的乙醚(15ml)溶液,搅拌1小时后加入碘甲烷(414μl,6.8mmol),反应液继续搅拌30min,20ml饱和氯化铵淬灭反应,乙醚萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为偕二甲基环丙烷类化合物15(25mg;步骤11和步骤12的总产率为44%,非对映体比例为1:1)。
[0133]
非对映体1体核磁数据:
[0134]1h nmr(400mhz,cdcl3)δ=5.53(d,j=1.6hz,1h),4.13
–
4.04(m,1h),4.02(d,j=9.0hz,1h),3.89(d,j=4.7hz,1h),3.23
–
3.11(m,1h),3.01(s,1h),2.42
–
2.35(m,1h),2.32(d,j=12.8hz,1h),2.02
–
1.86(m,2h),1.83(s,3h),1.79
–
1.65(m,3h),1.49
–
1.39(m,1h),1.12(s,3h),1.09(d,j=6.3hz,3h),0.96(s,3h),0.59(td,j=9.5,4.3hz,1h),0.33(dd,j=11.9,9.3hz,1h)。
[0135]
13
c nmr(100mhz,cdcl3)δ=219.5,131.1,126.2,79.6,73.8,60.7,47.5,41.1,38.7,33.4,32.2,30.7,29.7,28.8,21.7,21.5,18.4,18.1,15.0ppm。
[0136]
非对映体1高分辨质谱数据:
[0137]
hrms(esi):calcd for c
19h29o3+
[m+h]
+
305.2111,found 305.2115。
[0138]
非对映体2核磁数据:
[0139]1h nmr(400mhz,cdcl3)δ=5.64(d,j=4.9hz,1h),4.33(s,1h),3.84(d,j=6.8hz,1h),3.61(d,j=5.0hz,1h),3.32(d,j=8.5hz,1h),3.18
–
3.05(m,1h),2.59
–
2.44(m,1h),2.29(dd,j=11.6,2.0hz,1h),1.90(s,4h),1.88
–
1.78(m,2h),1.68(ddd,j=20.1,14.0,9.9hz,4h),1.41(ddd,j=22.9,15.0,9.0hz,1h),1.08(t,j=3.1hz,6h),0.94(s,3h),0.56(ddd,j=19.8,11.5,5.3hz,1h),0.23ppm(dd,j=11.4,9.5hz,1h)。
[0140]
13
c nmr(100mhz,cdcl3)δ=213.1,138.1,122.2,75.0,74.4,59.6,42.5,41.8,40.3,33.4,32.0,30.0,28.9,28.9,22.9,22.7,18.3,17.9,14.8ppm。
[0141]
非对映体2高分辨质谱数据:
[0142]
hrms(esi):calcd for c
19h29o3+
[m+h]
+
305.2111,found 305.2109。
[0143]
步骤13:
[0144]
[0145]
向上式所示的偕二甲基环丙烷类化合物15(25mg,0.082mmol)的二氯甲烷(5ml)和水(5ml)混合溶液中依次加入四甲基哌啶氧化物(2mg,0.008mmol),四丁基氯化铵(23mg,0.082mmol),碳酸氢钠(35mg,0.41mmol)和氯代丁二酰亚胺(22mg,0.16mmol),反应液搅拌2小时,10ml饱和碳酸氢钠溶液淬灭反应,二氯甲烷萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为偕二甲基二酮类化合物16(23.5mg,95%)。
[0146]
核磁数据:
[0147]1h nmr(400mhz,cdcl3)δ=5.42(dd,j=3.0,1.4hz,1h),4.00(s,1h),3.25(s,1h),3.19(ddd,j=10.6,8.1,6.5hz,1h),2.77(ddd,j=13.8,12.2,9.0hz,1h),2.32(d,j=11.6hz,1h),2.18(dd,j=18.8,8.8hz,1h),2.11
–
1.99(m,1h),1.98
–
1.88(m,2h),1.88
–
1.86(m,3h),1.75(ddd,j=20.3,14.6,9.6hz,2h),1.15(s,3h),1.07(d,j=6.4hz,3h),0.99(s,3h),0.59(td,j=9.4,4.1hz,1h),0.33ppm(dd,j=11.9,9.2hz,1h)。
[0148]
13
c nmr(100mhz,cdcl3)δ=216.1,215.2,133.8,120.0,73.6,58.4,50.0,39.9,37.8,34.8,30.2,29.6,28.7,25.9,21.5,21.3,18.3,17.4,15.1ppm。
[0149]
高分辨质谱数据:
[0150]
hrms(esi):calcd for c
19h27o3+
[m+h]
+
303.1955,found 303.1956。
[0151]
步骤14:
[0152][0153]
将偕二甲基二酮类化合物16(20mg,0.066mmol)的二氯甲烷(5ml)溶液冷却至0℃,依次缓慢加入三乙胺(91μl,0.66mmol)和三氟甲磺酸三甲基硅酯(48μl,0.264mmol),反应液在0℃搅拌30分钟,10ml饱和碳酸氢钠淬灭反应,二氯甲烷萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,所得物即为偕二甲基硅醚类化合物17(25mg)。
[0154]
步骤15:
[0155][0156]
将上式所示的偕二甲基硅醚类化合物17(25mg,0.066mmol)溶于乙腈(5ml),加入三氟醋酸钯(43mg,0.132mmol),反应液搅拌3小时后,硅胶过滤,减压浓缩,用硅胶柱层析法分离提纯产物得到白色粉末状固体,即为偕二甲基烯酮类化合物18(23mg,步骤14和步骤15的总产率为95%)。
[0157]
核磁数据:
[0158]1h nmr(400mhz,cdcl3)δ=7.38(d,j=5.6hz,1h),5.82(d,j=5.6hz,1h),5.59
–
5.42(m,1h),3.86(d,j=2.4hz,1h),3.66(dd,j=4.4,1.9hz,1h),3.13
–
2.97(m,1h),2.39
(dd,j=11.8,2.4hz,1h),1.98
–
1.87(m,1h),1.82
–
1.75(m,1h),1.74(t,j=1.6hz,3h),1.15(s,3h),1.03(d,j=6.4hz,3h),0.99(s,3h),0.65(td,j=9.3,4.9hz,1h),0.44(dd,j=11.7,9.3hz,1h),0.14ppm(s,9h)。
[0159]
13
c nmr(100mhz,cdcl3)δ=209.7,207.9,162.6,133.3,128.7,120.7,73.1,66.0,48.6,42.3,40.4,28.9,28.2,27.9,21.7,21.4,18.2,17.0,15.1,0.0(3c)ppm。
[0160]
高分辨质谱数据:
[0161]
hrms(esi):calcd for c
22h33
o3si
+
[m+h]
+
373.2193,found 373.2194。
[0162]
步骤16:
[0163][0164]
将上式所示的偕二甲基烯酮类化合物18(15mg,0.042mmol)的甲苯(4ml)溶液冷却至0℃,缓慢往里滴加甲基锂(1.6m乙醚溶液,53μl,0.084mmol),搅拌30min后,10ml饱和氯化铵溶液淬灭反应,乙醚萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到淡黄色油状物,即为偕二甲基烯丙醇类化合物19(13mg)。
[0165]
步骤17:
[0166][0167]
向上式所示的偕二甲基烯丙醇类化合物19(13mg,0.042mmol)的二氯甲烷(3ml)溶液中依次加入硅胶(18mg),乙酸钠(7mg,0.084mmol)和氯铬酸吡啶鎓盐(9mg,0.084mmol),混合液搅拌30分钟,随后加入盐酸(2m的水溶液,84μl,0.168mmol),搅拌30分钟,2ml饱和碳酸氢钠淬灭反应,二氯甲烷萃取,有机层用饱和食盐水洗涤和无水硫酸钠干燥,减压浓缩,用硅胶柱层析法分离提纯产物得到白色粉末状固体,即为pepluanol a(7.7mg;步骤16和步骤17的总产率为59%)。
[0168]
产物pepluanol a的核磁共振氢谱和核磁共振碳谱分别如图2和图3所示,本发明方法合成的产物与天然产物的nmr谱图数据一致。
[0169]
核磁数据:
[0170]1h nmr(400mhz,cdcl3)δ=5.77
–
5.70(brd,1h),5.69
–
5.65(brs,1h),3.96(brs,1h),3.92
–
3.86(brs,1h),3.56
–
3.44(m,1h),2.95(dd,j=11.8,2.5hz,1h),2.25(s,3h),2.01
–
1.92(m,1h),1.83(brs,3h),1.71(m,1h),1.52(d,j=4.1hz,1h),1.17(s,3h),0.98(s,3h),0.95(d,j=6.4hz,3h),0.61(td,j=9.2,5.2hz,1h),0.36ppm(dd,j=11.8,9.3hz,1h)。
[0171]
13
c nmr(100mhz,cdcl3)δ=207.3,203.4,181.8,134.6,125.1,122.7,73.6,71.7,
46.6,41.7,39.3,29.2,28.8,28.4,22.5,22.3,18.8,18.3,17.6,14.9ppm。
[0172]
高分辨质谱数据:
[0173]
hrms(esi):calcd for c
20h27o3+
[m+h]
+
315.1955,found 315.1957。
[0174]
以上所述的实施例对本发明的技术方案进行了详细说明,应理解的是以上所述的仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充或类似方式替代等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1