一种基于cfDNA碱基突变频率分布检测肝癌特异突变的方法
一种基于cfdna碱基突变频率分布检测肝癌特异突变的方法
技术领域
1.本发明涉及生物信息学技术领域,具体涉及cfdna中碱基突变频率分布的检测方法,及其在肿瘤预后评估中的应用。
背景技术:
2.癌症是危害人类健康的重大科学问题之一。据2020年统计数据显示,每年癌症新发病例可达1930万,死亡人数可达1000万,且发病率还处于逐年上升之中。目前,手术切除是多种肿瘤的主要治疗方式,但是肿瘤患者仍然面临着手术切除率不够理想,复发几率高的问题。术后微小残余病灶的存在是肿瘤手术后复发的主要原因,在手术后及时检测微小残余病灶,对于调整治疗方案,改善肿瘤患者预后,具有重要价值。目前,传统的影像学手段与血液标志物对于微小残余病灶的检测能力十分有限,难以解决临床上继续面对的预后评估问题。近年来得到高速发展的液态活检技术能够对患者的肿瘤负荷进行动态评估,具有较高的灵敏性,为肿瘤术后微小残余病灶的检测提供了新的工具与策略。
3.其中,基于血浆游离dna(circulating free dna)中循环肿瘤dna(circulating tumor dna)的基因组信息,使用测序技术追踪肿瘤基因组特征的动态肿瘤负荷评估策略已经成为临床应用热点领域之一。目前,国内外已经有多家不同临床研究机构同时开展了使用cfdna追踪检测患者术后肿瘤残余病灶以指导治疗策略优化的临床实验。在2020年,fda批准了第一个高通量测序技术的液态活检伴随治疗检测方案,在2021年,fda给予了signatera检测突破性医疗器械认定,肯定了其在评估微小残余病灶,监测预后中的价值。然而,目前在实体瘤特别是肝癌中,检测微小残余病灶还往往需要依赖组织取样先行获得的组织基因组信息。如何在不依赖组织的情况下,无创筛选肿瘤特异性突变并进行肿瘤负荷评估仍然是临床中需要解决的重大问题之一。
4.cfdna中包含了来自不同组织,不同细胞群的dna碎片。在患者的临床病程中,来自不同组织的dna碎片在cfdna中的占比将随着肿瘤负荷及患者的生理病理状态而发生动态性改变。因此,通过多时间点的cfdna样本,分析突变频率变化,就为筛选来自特定组织的突变提供了可能性。但是,目前使用时间序列cfdna样本进行肿瘤突变筛选,鉴定肿瘤负荷,评估微小残余病灶存在的方案还未得到建立。
技术实现要素:
5.为了解决上述技术难题,本发明开发了一种cfdna中碱基突变频率分布的检测方法,是对血液中的基因突变筛选策略,不需要依赖肿瘤组织取样,完全无创性的肿瘤突变检测方案,用于精确性筛选肿瘤来源突变。
6.本发明采用的技术方案是:一种基于cfdna碱基突变频率分布检测肝癌特异突变的方法,该方法包括以下步骤:
7.步骤1.从已提取的受试者的血浆中提取cfdna;
8.步骤2.对步骤1提取的cfdna的进行末端修复并在cfdna的3’端添加a碱基,在处理
后的cfdna两端连接测序带有分子标签umi的截短型y型接头,得到连接产物,并对连接产物进行磁珠纯化,用含有用于区分不同样本的index标签序列的引物进行扩增富集并磁珠纯化,加入带有生物素标记的探针进行杂交反应,用带有链和亲酶素标记的磁珠对文库进行洗脱,捕获生物素标记的核酸分子,并扩增和磁珠纯化,构建高通量测序文库,最后进行测序;
9.步骤3.对原始序列文件中的umi读段进行提取,分离每个读段两端各3个碱基的umi信息,将提取出的umi信息加到读段名字中进行保存后,将读段中umi信息从原始序列中切除,得到umi提取后的测序读段,将umi提取后的测序读段与人类参考基因组进行比对;
10.步骤4.用bwa提取所有比对到人类参考基因组同样位置的所有umi提取后的测序读段,若测序读段的umi信息相同,但碱基排列不一致,则去除该umi信息的测序读段;若多个测序读段的umi信息相同,且碱基排列一致,则标记为共通序列并保留;若只1个测序读段的umi信息相同,且碱基排列一致,则标记为孤立序列并保留;
11.步骤5.对孤立序列进行降噪后回收,与共通序列合并,形成最终的输出bam文件;
12.步骤6.使用ides算法将bam文件转化为碱基频率分布文件,然后使用人类参考基因组的参考数据集对碱基频率分布文件进行抛光,得到降噪之后的碱基频率分布;
13.步骤7.用pbmc的测序数据作为对照样本,对步骤6处理后的数据进行突变频率检测,去除假阳性突变,得到受试者的cfdna的突变频率分布。
14.进一步地,步骤7中突变频率检测中认定为突变的条件是:同时从正负双链对测序读段进行突变频率检测覆盖时有大于2个突变读段,或只从正链/负链对测序读段进行突变频率检测覆盖时有大于1个突变读段,同时突变频率大于-log(0.01)/depth,在对照样本中测序出的突变频率频率小于0.005。
15.进一步地,对孤立序列进行降噪后回收包括:计算所有只有孤立序列的人类参考基因组位置的碱基分布,基于二项分布评估测序噪声的大小,当对应孤立序列的碱基分布与人类参考基因组位置的主要碱基不一致,且碱基频率小于噪声时,将该孤立序列舍弃,否则则被回收。
16.进一步地,步骤7之后还包括:按步骤1~7检测肿瘤患者术前cfdna的突变频率分布和同种肿瘤组织样本的突变频率分布,鉴定其中重叠的部分,并比较重叠部分的突变频率分布,选取重叠部分的突变频率分布的下四分之一分位数,作为频率阈值。
17.进一步地,按步骤1~7检测肿瘤患者术后cfdna的突变频率分布,基于得到的频率阈值对肿瘤患者术前和术后cfdna的突变频率分布,及进行过滤,得到筛选后的突变频率分布,计算突变频率变化比率,并绘制峰图。
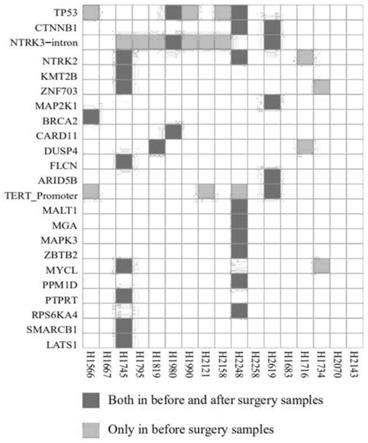
18.通过收集肝癌患者的手术前血液样本与手术后血液样本,分别提取cfdna进行高通量umi测序。经过高精度的序列处理与合并后,分别对每个血液样本进行突变检测。鉴定突变后,使用所有体细胞突变在手术前后两个时间点的频率变化对不同类型基因突变进行分群。然后对筛选到的肿瘤来源突变进行整合,筛选出的肿瘤来源特异突变可以进一步用于时间序列样本的肿瘤负荷动态评估。
19.本发明对肿瘤患者cfdna的突变谱进行过深入分析,首次发现,在肿瘤患者接受手术前后,其血浆cfdna的动态变化可以用于分离来自肿瘤的突变,且这些突变可以被用于评估肿瘤切除手术的效果。因此,只需要通过提取患者的术前术后cfdna,就可以无创性地对
肿瘤特异突变进行鉴定。
20.综上所述,利用肿瘤手术患者的术前与术后血浆样本对检测到的体细胞突变进行频率评估,可以筛选出来自肿瘤的特异突变。
21.本发明的有益效果主要体现在:本发明的技术方案剔除了受试者的cfdna的孤立序列噪声,突变背景噪声,假阳性突变,得到高精度的突变频率分布数据。高精度的突变频率分布数据还用频率阈值进行筛选,剔除了非肿瘤特异突变频率。
附图说明
22.图1.血浆中肿瘤组织特异性突变的分布;
23.图2.术前血浆中肿瘤组织特异性突变的频率分布;
24.图3.术前血浆肿瘤组织特异性突变在术后血浆中的频率分布;
25.图4.无组织样本术前术后突变频率比值的密度分布;
26.图5.阈值内外两组突变基因与肿瘤特异性突变基因的交叉情况,卡方检验:p值:0.00017;
27.图6.阈值上下两组突变reads长度150bp内的比例;
28.图7.分组与无复发生存时间(rfs)显著相关。
具体实施方式
29.下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此,本发明中所指的术语“术前”、“术后”是指切除肿瘤的手术前和手术后。
30.下述实施例中所用的实验材料,如无特殊说明,均为常规生化试剂商店购买得到。
31.材料与设备:
32.(1)qiaamp circulating nucleic acid提取试剂盒购自qiagen公司;
33.(2)链和亲酶素标记的磁珠购自integrated dna technologies(idt)公司,货号:1080589,链霉亲和素磁珠由磁微粒与高纯度链霉亲和素共价结合而成。磁珠可用于捕获生物素复合物,包括生物素标记的抗原、抗体和核酸。生物素-链霉亲和素的相互作用很强,非特异性结合率很低,使得被捕获的底物可满足后续实验要求;
34.(3)生物素标记的探针panel购自纳昂达(南京)生物科技有限公司,货号:1001111e,该探针panel涉及实体瘤研究中被广泛关注的578个基因,覆盖基因组约2.6mb区域;支持包括碱基替换、插入/缺失、基因重排、基因扩增、微卫星不稳定在内的多种变异信息的富集。
35.(4)consensuscruncher是一个抑制二代测序数据错误率的工具,通过唯一标识符unique molecular identifiers(umi)对同一dna模板上的read进行去重。
36.实施例1:
37.一、收集了100例肝细胞癌患者术前术后血浆样本与肿瘤组织样本,38例肝硬化患者的血浆样本与30健康志愿者的血浆样本。
38.二、根据收集入组肝细胞癌患者的术前术后血浆样本与肿瘤组织样本,从两者的随访血液样本中分别取8ml全血,使用两步离心法收集血浆,按照qiaamp circulating nucleic acid提取试剂盒的操作说明书提取血浆cfdna。
39.1、50ml离心管中加入3ml血浆、300μl proteinase k、2.4ml buffer acl(包含carrier rna),涡旋30s,60℃孵育30min。
40.2、取出离心管,加入5.4ml buffer acb,涡旋30s,冰上孵育5min。
41.3.将vacconnector,mini column以及20ml的tube extender依次插入到qiavac 24plus上。
42.4.将样品混合液加入tube extender中,打开真空泵,待样本过柱完毕后,关闭真空泵,移去20ml的tube extender,在mini column内依次加入600μl buffer acw1、750μl buffer acw2、750μl无水乙醇过柱清洗。
43.5、洗脱完毕后,拔出mini column放置在新的1.5ml ep管上,20000g离心3min,弃上清,开盖,置于孵育器中56℃孵育10min。
44.6、加30-50μl buffer ave到mini column中,常温孵育3min,20000g离心3min,收集cfdna样本。
45.7、利用qubit对cfdna的浓度进行精确定量,利用qsep 100毛细管电泳系统对cfdna片段大小进行检测。制备好的cfdna置于-80℃备用。
46.三、构建基因组文库(nanoprep
tm
dna文库构建试剂盒(for)搭配duplex seq adapters kit)
47.1、对cfdna的进行末端修复,并在3’端添加a碱基;
48.1)配置反应的混合液,如下表:
49.试剂用量cfdna10-20ngend repair&a-tailing buffer6μlend repair&a-tailing enzyme4μlh2o补足至50μl
50.与40μl的cfdna样本混匀后,放入pcr仪中反应条件如下:
51.温度时间20℃30min65℃30min10℃∞
52.2、cfdna两端连接测序带有分子标签(umi)的截短型y型接头,该接头含有随机分子标签序列。
53.1)配置反应的混合液,如下表:
54.试剂用量idt duplex adpater(15um)2μlligation buffer26μl总计28μl
55.与50μl的上一步的末端修复产物混匀后,最后加入2μl dnaligase,放入pcr仪反应条件如下:
56.温度时间
20℃15min4℃∞
57.2)连接产物纯化:
58.在连接产物中加入40μl nanoprep
tm
sp beads,混合均匀,常温孵育5-10min后放置在磁力架,待澄清后,弃去上清;加150μl 80%乙醇,室温孵育30s,漂洗磁珠两遍;去除残余的酒精,室温放置5-10min晾干磁珠;加入21μl nuclease free water,25℃孵育2min,磁力架吸附去除磁珠,转移上清20μl到新的0.2mlpcr管中,收集纯化的连接产物。
59.3.、用含有用于区分不同样本的index标签序列的引物进行扩增富集
60.1)配置反应的混合液,如下表:
61.试剂用量hifi pcr master mix,2x25μludi primer mix(5um)5μl总计30μl
62.与20μl的上一步纯化的连接产物混匀后,放入pcr仪反应条件如下:
[0063][0064]
2)pcr产物纯化在.pcr产物中加入50μl nanoprep
tm
sp beads,混合均匀,常温孵育5-10min后放置在磁力架,待澄清后,弃去上清;加150μl 80%乙醇,室温孵育30s,漂洗磁珠两遍;去除残余的酒精,室温放置5-10min晾干磁珠;加入21μl nuclease free water,25℃孵育2min,磁力架吸附去除磁珠,转移上清20μl到新的0.2mlpcr管中,收集纯化的pcr产物,进行qubit定量以及毛细管电泳。
[0065]
4.杂交反应
[0066]
1)取8-12个构建好的文库各500ng,混合成一个pool,加入5μl的cot human dna以及2μlblocker,真空干燥。
[0067]
2)杂交试剂配制
[0068]
试剂用量2x hybridization buffer8.5μlhybridization buffer enhancer2.7μlprobes4μlnuclease-free water1.8μl总计17μl
[0069]
用配制好的buffer重悬真空干燥后的产物,放入pcr仪孵育,条件如下:
[0070]
温度时间
95℃30s65℃16h
[0071]
5.进行杂交反应后用带有链和亲酶素标记的磁珠对文库进行洗脱,捕获生物素标记的核酸分子
[0072]
1)配制杂交洗脱buffer
[0073]
试剂用量h2o总计2x bead wash buffer160μl160μl320μl10x wash buffer 128μl252μl280μl10x wash buffer 216μl144μl160μl10x wash buffer 316μl144μl160μl10x stringent wash buffer32μl288μl320μl
[0074]
2)配制bead重悬mix
[0075]
试剂用量2x hybridization buffer8.5μlhybridization buffer enhancer2.7μlnuclease-free water5.8μl合计17μl
[0076]
3)杂交产物洗脱
[0077]
取50μl capture beads到1.5ml低吸附管,加入100μl bead wash buffer,用枪轻轻混匀,磁力架1min,去上清,加入配制好的17μl bead重悬mix重悬珠子,将准备好的capture beads加到杂交产物中,吹打混匀,65℃孵育45min捕获杂交文库。随后依次用配制好的用wash buffer1,stringent buffer,wash buffer 2,以及wash buffer 3,对捕获的杂交文库进行清洗,最后20μlnuclease-free water进行文库洗脱。
[0078]
6.pcr扩增杂交文库
[0079]
1)配制pcr试剂。
[0080]
试剂用量hifi pcr master mix,2x25μllibrary amplificationprimer mix(5um)5μl总计30μl
[0081]
与上一步的杂交洗脱产物混匀,进行pcr扩增,反应条件如下:
[0082][0083]
2)pcr产物纯化
[0084]
在.pcr产物中加入50μl nanoprep
tm
sp beads,混合均匀,常温孵育5-10min后放置在磁力架,待澄清后,弃去上清;加150μl 80%乙醇,室温孵育30s,漂洗磁珠两遍;去除残余的酒精,室温放置5-10min晾干磁珠;加入21μl nuclease free water,25℃孵育2min,磁力架吸附去除磁珠,转移上清20μl到新的0.2mlpcr管中,收集纯化的pcr产物,利用qubit对文库的浓度进行精确定量,利用qsep 100毛细管电泳系统对文库片段大小进行检测,片段主要集中为350bp左右。
[0085]
7、最后在illumina hiseq x ten平台上进行测序。
[0086]
四、使用consensuscruncher对原始序列文件中的umi(unique molecular identifiers分子标签)读段进行提取:
[0087]
umi模式设置为nnn,读取每个读段两端各3个碱基的umi信息,将提取出的umi信息加到读段名字中进行保存后,将读段中umi信息从原始序列中切除,得到umi提取后的测序读段,将umi提取后的测序读段与参考基因组hg19进行比对。
[0088]
五、继续使用consensuscruncher,将umi提取后的测序读段用bwa比对到hg19基因组上,记录位置信息后,进行umi共通序列的鉴定与孤立序列的分离。
[0089]
其中,共通序列指的是多个测序读段序列共同鉴定到比对位置,umi信息与碱基排序都完全一致的序列,孤立序列则是只有单个测序读段鉴定到的序列。
[0090]
具体分离过程如下:首先提取所有比对到基因组同样位置的所有umi测序读段,统计相应的umi信息的种类数目;判断所有比对到基因组同一位置同一umi信息的所有测序读段的碱基排列是否一致,若不一致,则该位置该umi信息的测序读段被视为引入pcr误差进行去除;当比对到基因组同一位置的同一umi信息的测序读段的碱基排列一致,且只有一个时,该测序序列被视为孤立序列,标记后进行保留,当比对到基因组同一位置的同一umi信息的测序读段的碱基排列一致,且大于一个时,这些测序序列被视为共通序列,标记后进行保留。其中,共通序列将直接被保留进入下一步分析;而孤立序列则应先行进行噪声处理:所有只有孤立序列支持的基因组位置的碱基分布将被计算,基于二项分布评估测序噪声的大小,当对应孤立序列的碱基分布与该位置的主要碱基不一致,且碱基频率小于噪声时,该孤立序列将被舍弃,否则则被回收,进入下一步分析。回收完毕的孤立序列与共通序列合并形成最终的bam文件。
[0091]
六、使用ides将bam文件转化为测序区域内部的碱基频率分布文件。
[0092]
ides算法基于每一个基因组位置,使用bam文件中的所有测序读段的序列信息。提取该位置的碱基分布。基于前期建立的hg19基因组的参考数据集的碱基在全基因组水平的分布对碱基频率分布文件进行抛光。抛光指代的是基于hg19基因组的碱基在全基因组水平的分布,构建分布模型,根据该模型对测序读段的候选突变进行假设检验,判断该候选突变的真实性,估算突变碱基错误检出的概率,若突变为背景错误,予以去除,抛光之后,碱基分布文件中的背景错误率将得到下降。
[0093]
七、最后使用样本间频率分布差异进行高精度突变检测。
[0094]
构建pbmc(外周血单个核细胞)的基因文库,按步骤四~六获得测序数据,作为对照样本,进行术前血浆样本的突变频率检测,要求突变位点符合下列要求:为双端(即同时有正负双链读段支持)序列覆盖时有大于2个突变读段支持,为单端序列(即只有正链或负链的读段支持)覆盖时有大于1个突变读段支持,同时突变频率大于-log(0.01)/depth,在
对照样本中测序出的突变频率小于0.005,以去除假阳性结果。
[0095]
八、对术前检测到的突变与来自肿瘤组织的突变进行交叉比对,鉴定其中重叠的部分,并比较重叠部分(在血液与组织中都能检出的突变,即血液中检测到的肿瘤特异突变)的突变频率分布。
[0096]
重叠的突变的频率分布如图2所示。该图显示,肿瘤特异突变在血液中的频率分布存在特定模式,相应的,血液中的非肿瘤特异突变频率则更多处于低频区域。因此,选取血液中肿瘤特异突变频率分布的下四分之一分位数,0.02611作为突变频率筛选的依据。这一频率阈值将作为在不依赖组织情况下过滤突变的第一个条件。
[0097]
将所有数值按大小顺序排列并分成四等份,处于三个分割点位置的得分就是四分位数。最小的四分位数称为下四分位数。
[0098]
九、基于上一步确定的频率阈值0.02611对术前检测到的所有突变进行过滤,得到筛选后的突变作为肿瘤来源突变的备选。按步骤三~七对术后血液样本对大于频率阈值的这些位点的突变频率进行提取,术前血浆肿瘤组织特异性突变在术后血浆中的频率分布如图3所示。
[0099]
十、计算比较手术前(图2)和术后(图3)突变频率变化的比率,绘制峰图如图4所示。该图显示,按照突变频率变化情况,对峰图进行观测,选定能明确分离两个不同突变峰的界限值作为诊断阈值,该诊断阈值处于两个突变峰之间下陷的最低点,术前检测到的突变可以分为明确的两群,其中出现显著下降的一群应反映了肿瘤切除手术对肿瘤负荷的影响,应为肿瘤特异突变;另外一群则应为其他来源(即非肿瘤特异来源)突变。
[0100]
十一、提取该群突变进行基因重叠分析(图5)与长度分布分析(图6),结果证实这一群突变具有典型的肿瘤来源突变的特征。因此该群突变可被认为是肿瘤来源的特异性突变。
[0101]
十三、基于该群突变在术后样本的有无判断肝细胞癌患者肿瘤的微小残余病灶,并使用r进行km生存分析(图7)。结果显示,具有微小残余病灶的肝细胞癌患者预后显著差于没有检测出微小残余病灶的患者。以上结果突出显示基于手术前后体细胞突变的频率变化可以在不依赖肿瘤组织的前提下,筛选肿瘤来源突变,并以此鉴定肿瘤微小残余病灶。
[0102]
实施例2:
[0103]
取病人手术前和术后一月的血浆样本按实施例1的方法进行ctdna检测分析,存在突变术前术后频率变化率大于诊断阈值(0.2)的突变。经过分析这些突变在基因上的分布与序列长度分布,发现突变基因包括与肿瘤发生相关的重要基因,因此为肿瘤相关突变。经过分析,发现肿瘤相关突变在术后仍然存在,判定为有微小残余病灶。后经随访发现,该病人在术后底84天出现影像学复发。
[0104]
实施例3:
[0105]
取病人手术前和术后一月的血浆样本按实施例1的方法进行ctdna检测分析,存在突变术前术后频率变化率大于阈值(0.2)的突变。基因分布与序列长度分布分析支持该群突变为肿瘤特异突变。在术后样本中,该群突变的频率全部为0,判定为无微小残余病灶。后经随访发现,该病人在术后一年内未复发。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1