一种环状RNA分子及应用的制作方法
一种环状rna分子及应用
技术领域
1.本发明涉及一种环状rna分子,具体涉及一种能表达肺癌通用型新生抗原的环状rna分子及应用。
背景技术:
2.肺癌是导致人类死亡率最高的恶性肿瘤。2020年中国癌症死亡人数300万,肺癌死亡人数遥遥领先,高达71万,占癌症死亡总数的23.8%。这与我国大部分肺癌患者在确诊时已为中晚期,预后较差有关。尽管近年来肺癌的分子靶向治疗和免疫治疗技术发展较快,但并未显著延长患者的总生存时间。因为接受靶向治疗或免疫治疗的患者最终会产生治疗抵抗,而失去效果,肿瘤会再次复发。对于晚期患者来说,医生不会谈论治愈癌症,他们希望有药物能将这种棘手的疾病变为慢性疾病,延长患者生存期,即便无法治愈,也可以实现长期带瘤生存,将肺癌控制为像高血压,糖尿病一样的慢性病。
3.如果有一种癌症疫苗能够成功触发强烈的免疫应答并且在适合的人群中使用,就可以显著改善肺癌存活。临床研究显示,三种正在研究的肺癌疫苗如tecemotide,tg4010和belagenpumatucel-l暂时不符合主要终点。癌症疫苗的历史经历了许多失败,但癌症疫苗的研发一直是医学研究领域激烈探索的热点。
4.2020年4月1日,ose immunotherapeutics公司宣布其新型抗癌疫苗tedopi在非小细胞肺癌的ⅲ期临床试验取得了阳性结果,入组的所有患者在免疫检查点抑制剂pd-1耐药或失败后,二线或三线使用tedopi疫苗,一年的总生存率高达46%。也就是说,tedopi为晚期及pd-1治疗耐药后走投无路的患者提供了新的选择和希望。但是,tedopi仅限于hla-a2阳性(约占45%)患者。
5.mrna是一种天然存在的分子,带有人类细胞的“蓝图”,可以产生靶标蛋白或免疫原,激活体内免疫反应。mrna疫苗是将含有编码抗原蛋白的mrna导入人体,直接进行翻译,状成相应的抗原蛋白,从而诱导机体产生特异性免疫应答,从而使机体获得免疫保护。在新冠疫情爆发和病毒不断变异的背景下,mrna 技术在默默发展多年后,终于开始从实验室研究全面走向临床研究和应用。但是mrna存在rna酶水解耐受性差的缺点。目前发现作为“线性mrna2.0”的环状mrna具有较好的rna酶水解耐受性。与此同时,环状rna因其独特的构象,使其可以在生物体内非常稳定的存在,从而能够长时间的持续表达蛋白,使长效性的蛋白替代或者蛋白过表达的rna药物成为可能,实现mrna无法实现的药物需求。
6.所以,非常有必要开发环状rna分子作为通用型肺癌疫苗,扩大肺癌疫苗治疗的患者范围,进一步提高肺癌患者生存率,给患者本人、患者家庭和社会带来更多的癌症治疗受益。
技术实现要素:
7.发明要解决的问题本发明的目的在于提供一种环状rna分子。所述环状rna分子能够编码表达通用型
肺癌抗原多肽蛋白,诱导免疫应答,是一类潜在的肿瘤免疫类药物,有益于不同类型肺癌的预防和治疗,可以为肺癌病患及其家庭和社会带来免疫治疗收益。
8.为实现前述发明目的,本发明采用的技术方案包括:本发明实施例提供了一种环状rna分子,所述环状rna分子的ires元件含有以下a1-a4所述的其中一项序列:a1 seq id 1-3任一序列所组成的组中的一种或多种序列的核苷酸序列;a2 seq id 1-3任一序列所示的序列的反向互补序列的核苷酸序列;a3在高严格性杂交条件或非常高严格性杂交条件下,能够与a1或a2所示的核苷酸序列杂交的序列的反向互补序列;a4与a1或a2所示的核苷酸序列具有至少80%,81%,82%,83%,84%,85%,86%,87%,88%,89%,90%,91%,92%,93%,94%,95%,96%,97%,98%或99%的序列同一性的序列。
9.进一步的,所述环状rna分子能够表达下列通用型肺癌肿瘤新生抗原,其包括下列多肽中的任一种或多种的组合:具有seq id 4所示的序列或其保守变异型序列的多肽;具有seq id 5所示的序列或其保守变异型序列的多肽;具有seq id 6所示的序列或其保守变异型序列的多肽;具有seq id 7所示的序列或其保守变异型序列的多肽;具有seq id 8所示的序列或其保守变异型序列的多肽;具有seq id 9所示的序列或其保守变异型序列的多肽;具有seq id 10所示的序列或其保守变异型序列的多肽;具有seq id 11所示的序列或其保守变异型序列的多肽;具有seq id 12所示的序列或其保守变异型序列的多肽;具有seq id 13所示的序列或其保守变异型序列的多肽;具有seq id 14所示的序列或其保守变异型序列的多肽;具有seq id 15所示的序列或其保守变异型序列的多肽;具有seq id 16所示的序列或其保守变异型序列的多肽;具有seq id 17所示的序列或其保守变异型序列的多肽;具有seq id 18所示的序列或其保守变异型序列的多肽;具有seq id 19所示的序列或其保守变异型序列的多肽;具有seq id 20所示的序列或其保守变异型序列的多肽;具有seq id 21所示的序列或其保守变异型序列的多肽;具有seq id 22所示的序列或其保守变异型序列的多肽;具有seq id 23所示的序列或其保守变异型序列的多肽;具有seq id 24所示的序列或其保守变异型序列的多肽;具有seq id 25所示的序列或其保守变异型序列的多肽;具有seq id 26所示的序列或其保守变异型序列的多肽;具有seq id 27所示的序列或其保守变异型序列的多肽;具有seq id 28所示的序列或其保守变异型序列的多肽;
具有seq id 29所示的序列或其保守变异型序列的多肽;具有seq id 30所示的序列或其保守变异型序列的多肽;具有seq id 31所示的序列或其保守变异型序列的多肽;具有seq id 32所示的序列或其保守变异型序列的多肽;具有seq id 33所示的序列或其保守变异型序列的多肽;具有seq id 34所示的序列或其保守变异型序列的多肽;具有seq id 35所示的序列或其保守变异型序列的多肽;具有seq id 36所示的序列或其保守变异型序列的多肽;具有seq id 37所示的序列或其保守变异型序列的多肽;具有seq id 38所示的序列或其保守变异型序列的多肽;具有seq id 39所示的序列或其保守变异型序列的多肽;具有seq id 40所示的序列或其保守变异型序列的多肽;具有seq id 41所示的序列或其保守变异型序列的多肽;具有seq id 42所示的序列或其保守变异型序列的多肽;具有seq id 43所示的序列或其保守变异型序列的多肽;具有seq id 44所示的序列或其保守变异型序列的多肽;具有seq id 45所示的序列或其保守变异型序列的多肽;具有seq id 46所示的序列或其保守变异型序列的多肽;具有seq id 47所示的序列或其保守变异型序列的多肽;具有seq id 48所示的序列或其保守变异型序列的多肽;具有seq id 49所示的序列或其保守变异型序列的多肽;更进一步的,所述保守变异型序列为原始序列中的一个或多个氨基酸缺失、取代或插入而得,其具有至少80%,81%,82%,83%,84%,85%,86%,87%,88%,89%,90%,91%,92%,93%,94%,95%,96%,97%,98%或99%的原始序列同一性的序列。
10.本发明实施例还提供了一种重组表达载体,所述重组表达载体包含有本发明前述实施例所述的环状rna分子。
11.本发明实施例还提供了一种重组宿主细胞,其包含有本发明前述实施例所述的环状rna分子;进一步的,所述重组宿主细胞,其包含有本发明前述实施例所述的重组表达载体;本发明实施例还提供了一种在制备蛋白中应用的对象,其包含有本发明前述实施例所述的环状rna分子;进一步的,所述在制备蛋白中应用的对象,其包含有本发明前述实施例所述的重组表达载体;进一步的,所述在制备蛋白中应用的对象,其包含有本发明前述实施例所述的重组宿主细胞。
12.本发明实施例还提供了一种药物组合物,其包含有本发明前述实施例所述的肿瘤新生抗原多肽;进一步的,所述的药物组合物,其包含有本发明前述实施例所述的环状rna分子;进一步的,所述的药物组合物,其包含有本发明前述实施例所述的重组表达载体;
进一步的,所述的药物组合物,其包含有本发明前述实施例所述的重组宿主细胞。
13.本发明实施例还提供了一种制备蛋白的方法,其包含有本发明前述实施例所述的肿瘤新生抗原多肽表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的环状rna分子表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的重组表达载体表达目标蛋白的过程;进一步的,所述的制备蛋白的方法,其包含有本发明前述实施例所述的重组宿主细胞表达目标蛋白的过程。
14.本发明实施例还提供了一种预防或治疗肺癌的方法,其包括向受试者施用本发明前述实施例所述的环状rna分子;进一步的,所述预防或治疗肺癌的方法,其包括向受试者施用本发明前述实施例所述的肿瘤新生抗原多肽;进一步的,所述预防或治疗肺癌的方法,其包括向受试者施用本发明前述实施例所述的重组表达载体;进一步的,所述预防或治疗肺癌的方法,其包括向受试者施用本发明前述实施例所述的重组宿主细胞。
15.本发明的积极效果如下:本发明提供了一种环状rna分子。所述环状rna分子能表达肺癌通用型肿瘤新生抗原,诱导免疫应答,是一类潜在的肿瘤免疫类药物,有益于肺癌的预防和治疗,可以为肺癌病患及其家庭和社会带来免疫治疗收益。
附图说明
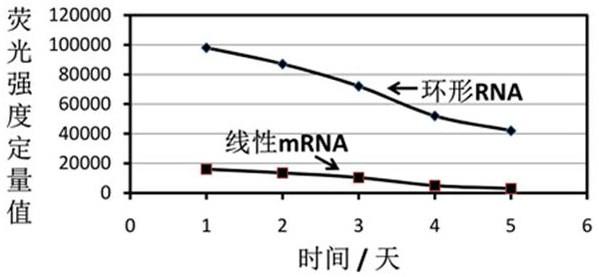
16.图1为含有seq id 1所述的其中一项序列ires介导状成的环状mrna和相对应的线性mrna在293t细胞中转染1-5天后对egfp蛋白表达的荧光强度比较。
17.图2为含有seq id 2所述的其中一项序列ires介导状成的环状mrna和相对应的线性mrna在293t细胞中转染1-5天后对egfp蛋白表达的荧光强度比较。
18.图3为含有seq id 3所述的其中一项序列ires介导状成的环状mrna和相对应的线性mrna在293t细胞中转染1-5天后对egfp蛋白表达的荧光强度比较。
具体实施方式
19.鉴于现有技术中的不足,本案发明人在长期研究和大量实践的基础上,得以提出本发明的技术方案。
20.如在权利要求和说明书中所使用的,词语“具有”、“包括”或“含有”是指包括在内的或开放式的,并不排除额外的、未引述的元件或方法步骤。
21.如在说明书中所使用的,术语“多肽”在本发明中为氨基酸聚合物。所述氨基酸聚合物可以是线状或分支的。进一步地,所述氨基酸聚合物包括修饰的氨基酸或由非氨基酸隔断的状式。更进一步地,所述氨基酸聚合物包括二硫键状成、糖基化、脂质化、乙酰化、磷酸化或以标记组分缀合的氨基酸聚合物。
22.如本发明所使用的,术语“环状rna”是指一种呈封闭状的环状rna分子。所述环状rna分子主要由外显子、ires元件、蛋白编码区和间隔区构成。由于本发明所使用的环状rna具有蛋白翻译活性,所述环状rna又可称为“环状mrna”。
23.如本发明所使用的,术语“一个或多个氨基酸缺失、取代或插入”中“缺失”是指去除占据某一位置的氨基酸,“取代”是指用不同氨基酸置换占用一个位置的核苷酸或氨基酸,“插入”是指在邻接并且紧随占据位置的氨基酸之后添加氨基酸。
24.如本发明所使用的,术语“保守变异”是指可正常维持蛋白质功能的保守变异。所述保守变异包括保守置换。所述保守置换是指在置换部位为芳香族氨基酸的情况下,在 phe、trp、tyr间相互置换的突变;在置换部位为疏水性氨基酸的情况下,在leu、ile、val间相互置换的突变;在为极性氨基酸的情况下,在gln、asn间相互置换的突变;在为碱性氨基酸的情况下,在lys、arg、his间相互置换的突变;在为酸性氨基酸的情况下,在asp、glu间相互置换的突变;在为具有羟基的氨基酸的情况下,在ser、thr间相互置换的突变;其他被视作保守置换的置换, ala向ser或thr的置换、arg向gln、his或lys的置换、asn向glu、gln、lys、his或asp的置换、asp向asn、glu或gln的置换、cys向ser或ala的置换、gln向asn、glu、lys、his、asp或arg的置换、glu向gly、asn、gln、lys或asp的置换、gly向 pro的置换、his向asn、lys、gln、arg或tyr的置换、ile向leu、met、val或phe的置换、leu向 ile、met、val或phe的置换、lys向asn、glu、gln、his或arg的置换、met向ile、leu、val或phe 的置换、phe向trp、tyr、met、ile或leu的置换、ser向thr或ala的置换、thr向ser或ala的置换、trp向phe或tyr的置换、tyr向his、phe或trp的置换、及val向met、ile或leu的置换。进一步地,所述保守突变涵盖因于基因所来源的个体差异、株、种的差异等天然产生的突变。
25.如本发明所使用的,术语“序列同一性”和“同一性百分比”是指两个或更多个多核苷酸或多肽之间相同(即同一)的核苷酸或氨基酸的百分比。
26.如本发明所使用的,术语“反向互补序列”是指和原始多核苷酸序列方向相反但互补的序列。
27.在本发明中,术语“载体”是指dna构建体。所述dna构建体含有合适的控制序列。所述控制序列可操作地连接dna序列,实现在合适的宿主中表达目的基因。
28.如在本发明所使用的,术语“重组表达载体”是指用于表达例如编码所需多肽的多核苷酸的dna结构。所述重组表达载体包括启动子和增强子等对基因表达具有调控作用的遗传元素的集合、能转录成mrna并翻译成蛋白质的结构或编码序列、能适当的转录和翻译起始和终止序列的转录亚单位。所述重组表达载体可以以任何合适的方式构建。在本发明中,重组表达载体的载体可以包括质粒、病毒、噬菌体和转座子在内的任何载体。
29.如在本发明所使用的,术语“抗原”是指引发免疫应答的分子。所述免疫应答是指可能涉及抗体产生和或特异性免疫细胞的活化。所述抗原包括基本上所有的蛋白质或肽在内的任何大分子。
30.如在本发明所使用的,术语“宿主细胞”是指易于用包含本发明的环状rna或重组表达载体转化、转染、转导等的任何细胞类型。
31.如在本发明所使用的,术语“重组宿主细胞”是指通过转化来实现的宿主细胞,包括导入环状rna或重组表达载体后不同于亲本细胞的宿主细胞。所述宿主细胞包括原核细胞或真核细胞,或能够导入本发明的环状rna或重组表达载体的细胞。
32.如在本发明所使用的,术语“转化、转染、转导”是指将外源性的dna导入宿主的过程。所述转化、转染、转导的方法包括任何将核酸导入细胞的方法。所述方法包括但不限于电穿孔法、磷酸钙(capo
4 )沉淀法、氯化钙(cacl
2 )沉淀法、微注射。法、聚乙二醇(peg)法、deae-葡聚糖法、阳离子脂质体法以及乙酸锂-dmso法。
33.如在本发明所使用的,术语“治疗”是指在罹患疾病之后,使受试者在接触本发明的菌株和/或巨噬细胞或者含有其的药物组合物后,相比受试者不接触时,使受试者的该疾病症状减轻,但并不意味着必需完全抑制受试者该疾病的症状。
34.在一些实施方案中,“本发明的药物组合物”是指含有本发明的菌株和/或巨噬细胞的药物组合物。
35.如在本发明所使用的,术语“预防”是指在罹患疾病之前,通过使受试者接触本发明的药物组合物,从而与受试者不接触时相比减轻罹患疾病后的症状,并不意味着必需完全抑制受试者患病。
36.如在本发明所使用的,术语“个体”、“患者”或“受试者”包括哺乳动物。所述哺乳动物包括但不限于,家养动物,灵长类动物,兔,以及啮齿类动物。
37.如在本发明所使用的,术语“高严格条件”是指根据标准dna印迹程序,对于长度为至少100个核苷酸的探针而言,在42摄氏度处在5x sspe、0.3%sds、200微克/毫升剪切并变性的鲑精dna和50%甲酰胺中预杂交和杂交12至24小时。最后在65摄氏度处使用2xssc、0.2%sds将载体材料洗涤三次,每次15分钟。
38.如在本发明所使用的,术语“非常高严格条件”是指根据标准dna印迹程序,对于长度为至少100个核苷酸的探针而言,在42摄氏度处在5x sspe、0.3%sds、200微克/毫升剪切并变性的鲑精dna和50%甲酰胺中预杂交和杂交12至24小时。最后在70摄氏度处使用2x ssc、0.2%sds将载体材料洗涤三次,每次15分钟。
39.除非另外定义或由背景清楚指示,否则在本公开中的全部技术与科学术语具有如本公开所属领域的普通技术人员通常理解的相同含义。
40.下面的实施例是对本发明的技术方案、其实施过程及原理等作进一步的解释说明。
41.下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
42.下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
43.实施例1 ires介导形成的环状mrna在293t细胞中的表达第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,egfp编码区 (seq id 55),3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(279.3纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化
要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(134.6纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnase i在37摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(1064.5纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(521.7纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到90%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,定量检测细胞转染后1-5天的荧光强度(图 1),较高的荧光强度表明本专利提供的ires介导状成的环状mrna具有较好的egfp蛋白表达能力,比相对应的ires介导的线性mrna对egfp的表达更强且更持久(图 1)。
44.实施例2 ires介导形成的环状mrna在293t细胞中的表达第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,egfp编码区 (seq id 55),3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,217rpm活化3.2小时后,取活化菌液在37.2摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(280.4纳克每微
升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(143.8纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.8摄氏度孵育2小时,然后加入dnase i在37.1摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(1150.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.8摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(491.9纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到90%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,定量检测细胞转染后1-5天的荧光强度(图 2),较高的荧光强度表明本专利提供的ires介导状成的环状mrna具有较好的egfp蛋白表达能力,比相对应的ires介导的线性mrna对egfp的表达更强且更持久(图 2)。
45.实施例3 ires介导形成的环状mrna在293t细胞中的表达第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,egfp编码区 (seq id 55),3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,221rpm活化3.2小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(278.4纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(152.2纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnase i在37摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(1208.7纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.7摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(437.1纳克每微升); 第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到80%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,定量检测细胞转染后1-5天的荧光强度(图 3),较高的荧光强度表明本专利提供的ires介导状成的环状mrna具有较好的egfp蛋白表达能力,比相对应的ires介导的线性mrna对egfp的表达更强且更持久(图 3)。
46.实施例4 编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证
第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,具有seq id 4所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.8摄氏度,220rpm活化2.9小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(321.5纳克每微升); 第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(202.4纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2小时,然后加入dnase i在37.1摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(653.9纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.6摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(312.5纳克每微升); 第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存; 第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为38次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到80%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;
第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为15.2纳克每毫升,33.6纳克每毫升,65.5纳克每毫升,47.8纳克每毫升, 22.1纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 4所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为3.1纳克每毫升,6.4纳克每毫升,13.5纳克每毫升,8.5纳克每毫升,4.2纳克每毫升)具有更出色的seq id 4所示氨基酸序列的抗原多肽蛋白的表达性能。
47.实施例5 编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证 第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,具有seq id 6所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,219rpm活化3小时后,取活化菌液在37.1摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(345.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(244.8纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2小时,然后加入dnase i在36.9摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(521.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.5摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(267.3纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存; 第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为36次); 4摄氏度
放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到70%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为21.3纳克每毫升,36.1纳克每毫升,53.4纳克每毫升,30.2纳克每毫升, 12.6纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 6所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为5.5纳克每毫升,8.4纳克每毫升,16.9纳克每毫升,10.5纳克每毫升,3.9纳克每毫升)具有更出色的seq id 6所示氨基酸序列的抗原多肽蛋白的表达性能。
48.实施例6编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,具有seq id 8所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3.1小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(444.5纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(290.2纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育2小时,然后加入dnase i在37.1摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(572.9纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.5摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(267.4纳克每微升);
第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为38次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到85%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为33.4纳克每毫升,47.2纳克每毫升,68.1纳克每毫升,50.3纳克每毫升, 24.5纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 8所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为6.1纳克每毫升,8.2纳克每毫升,12.1纳克每毫升,9纳克每毫升,4.7纳克每毫升)具有更出色的seq id 8所示氨基酸序列的抗原多肽蛋白的表达性能。
49.实施例7编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,具有seq id 10所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(297.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(264.2纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育2小时,然后加入dnase i在37.1摄氏度消化15分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并
同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(406.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在54.5摄氏度加热15分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(278.3纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为37次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2.5天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到75%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为65.6纳克每毫升,81.9纳克每毫升,120.7纳克每毫升,80.2纳克每毫升, 44.9纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 10所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为9.7纳克每毫升,13.6纳克每毫升,23.7纳克每毫升,11纳克每毫升,6.6纳克每毫升)具有更出色的seq id 10所示氨基酸序列的抗原多肽蛋白的表达性能。
50.实施例8编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,具有seq id 12所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,221rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(405.8纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯
化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(262.9纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2小时,然后加入dnase i在37.1摄氏度消化25分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(531.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在56.5摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(246.3纳克每微升; 第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37.1摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为30-40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列,说明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2-3天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到85%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为38.2纳克每毫升,51.1纳克每毫升,70.2纳克每毫升,55.8纳克每毫升, 30.9纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 12所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为6.2纳克每毫升,9.4纳克每毫升,13.8纳克每毫升,10纳克每毫升,4.4纳克每毫升)具有更出色的seq id 12所示氨基酸序列的抗原多肽蛋白的表达性能。
51.实施例9编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,具有seq id 14所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用
于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(367.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(255.4纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育3小时,然后加入dnase i在36.9摄氏度消化21分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(492.3纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(278.8纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为36次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔3天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到83%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为90.9纳克每毫升,134.4纳克每毫升,170.7纳克每毫升,125.3纳克每毫升, 62.5纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 14所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分
别为12.1纳克每毫升,19.3纳克每毫升,33.2纳克每毫升,17纳克每毫升,9.5纳克每毫升)具有更出色的seq id 14所示氨基酸序列的抗原多肽蛋白的表达性能。
52.实施例10编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,具有seq id 17所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,220rpm活化3.7h后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(270.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(222.6纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育3小时,然后加入dnase i在36.9摄氏度消化20分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(384.7纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(272.4纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为37次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示,产物包含连接后的第二外显子和第一外显子序列。其说明:环状 rna已成功制备;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37
摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔3天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到80%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为22.2纳克每毫升,34.1纳克每毫升,51.6纳克每毫升,31.7纳克每毫升, 20.3纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 17所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为3.5纳克每毫升,6.6纳克每毫升,9.5纳克每毫升,6.2纳克每毫升,3.2纳克每毫升)具有更出色的seq id 17所示氨基酸序列的抗原多肽蛋白的表达性能。
53.实施例11编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证 第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,具有seq id 20所示氨基酸序列的肿瘤抗原多肽编码区,3’间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在37.1摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(286.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(233.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnase i在36.9摄氏度消化20分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(342.7纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热18分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(202.1纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1
微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到80%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为35.1纳克每毫升,44.7纳克每毫升,59.8纳克每毫升,51.3纳克每毫升, 27.9纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 20所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为5.3纳克每毫升,7.8纳克每毫升,12.4纳克每毫升,6.9纳克每毫升,4.6纳克每毫升)具有更出色的seq id 20所示氨基酸序列的抗原多肽蛋白的表达性能。
54.实施例13编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,具有seq id 23所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(302.1纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(262.7纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.5小时,然后加入dnase i在37.1摄氏度消化20分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(485.6纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混
合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(248.4纳克每微升); 第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热18分钟,随后在85摄氏度加热8秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为39次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明说明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔3天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到88%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为117.2纳克每毫升,240.3纳克每毫升,346.9纳克每毫升,211.4纳克每毫升, 107.6纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 23所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为15.4纳克每毫升,26.4纳克每毫升,34.2纳克每毫升,21.8纳克每毫升,11.5纳克每毫升)具有更出色的seq id 23所示氨基酸序列的抗原多肽蛋白的表达性能。
55.实施例14编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,具有seq id 26所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(248.4纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(220.5纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为
20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnase i在37摄氏度消化25分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(374.8纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(260.1纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20分钟,随后在85摄氏度加热9秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到90%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为66.6纳克每毫升,84.1纳克每毫升,73.3纳克每毫升,52.6纳克每毫升, 36.5纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 26所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为15.4纳克每毫升,12.1纳克每毫升,17.7纳克每毫升,27.2纳克每毫升,10.1纳克每毫升)具有更出色的seq id 26所示氨基酸序列的抗原多肽蛋白的表达性能。
56.实施例15编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,具有seq id 29所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37.1摄氏度,219rpm活化3.2小时后,取活化菌液在36.9摄氏度,219rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(305.7纳克每微
升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(282.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2.1小时,然后加入dnase i在37.1摄氏度消化21分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(555.8纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在56摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(265.8纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热19分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存; 第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于36.9摄氏度,5%co2培养箱中培养。待细胞达到88%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为37.8纳克每毫升,47.4纳克每毫升,66.6纳克每毫升,50.1纳克每毫升, 29.4纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 29所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为6.2纳克每毫升,9.5纳克每毫升,15.3纳克每毫升,8.4纳克每毫升,4.7纳克每毫升)具有更出色的seq id 29所示氨基酸序列的抗原多肽蛋白的表达性能。
57.实施例16编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证
第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,具有seq id 32所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(391.7纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在36.9摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(276.8纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnase i在36.9摄氏度消化20分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(337.9纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热25分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(241.4纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热20.5分钟,随后在85摄氏度加热12秒,最后在4摄氏度保存; 第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为35次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到83%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;
第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为60.5纳克每毫升,78.3纳克每毫升,90.9纳克每毫升,78.1纳克每毫升, 47.2纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 32所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为7.5纳克每毫升,12.1纳克每毫升,19.8纳克每毫升,8.2纳克每毫升,5.1纳克每毫升)具有更出色的seq id 32所示氨基酸序列的抗原多肽蛋白的表达性能。
58.实施例17 编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,具有seq id 35所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(277.9纳克每微升); 第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(204.2纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2.1小时,然后加入dnase i在37摄氏度消化22分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(384.9纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在56.3摄氏度加热21分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(254.5纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20 分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为38次); 4摄氏度
放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到83%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为78.1纳克每毫升,116.4纳克每毫升,220.7纳克每毫升,112.3纳克每毫升, 66.8纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 35所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为11.4纳克每毫升,23.5纳克每毫升,39.6纳克每毫升,18.7纳克每毫升,9.9纳克每毫升)具有更出色的seq id 35所示氨基酸序列的抗原多肽蛋白的表达性能。
59.实施例18 编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,具有seq id 38所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(364.8纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(244纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnase i在37摄氏度消化25分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(484.2纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热20分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(272.6纳克每微升);
第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37.1摄氏度加热20.2分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存; 第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为40次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于36.9摄氏度,5%co2培养箱中培养。待细胞达到90%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为39.4纳克每毫升,56.8纳克每毫升,80.3纳克每毫升,52.1纳克每毫升,27.2纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 38所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为6.7纳克每毫升,10.2纳克每毫升,19.5纳克每毫升,12.4纳克每毫升,5.8纳克每毫升)具有更出色的seq id 38所示氨基酸序列的抗原多肽蛋白的表达性能。
60.实施例19编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,具有seq id 41所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(248.3纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(218.6纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2小时,然后加入dnase i在37摄氏度消化25分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时
通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(356.8纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热25分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(232.1纳克每微升); 第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20 分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为36次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到83%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为20.8纳克每毫升,35.2纳克每毫升,60.7纳克每毫升,42.3纳克每毫升,18.4纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 41所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为4.1纳克每毫升,6.9纳克每毫升,12.1纳克每毫升,8.5纳克每毫升,3.5纳克每毫升)具有更出色的seq id 41所示氨基酸序列的抗原多肽蛋白的表达性能。
61.实施例20 编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 1所示核苷酸序列的ires元件,具有seq id 44所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在37摄氏度,220rpm活化3小时后,取活化菌液在37摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(373纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化
要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(292.1纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37摄氏度孵育2.5小时,然后加入dnase i在36.9摄氏度消化25分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(544.4纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热25分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(256.4纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20 分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为37次);4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到89%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为51.3纳克每毫升,65.8纳克每毫升,74.9纳克每毫升,55.2纳克每毫升,38.5纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 44所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为8.8纳克每毫升,12.5纳克每毫升,19.6纳克每毫升,11.8纳克每毫升,5.2纳克每毫升)具有更出色的seq id 44所示氨基酸序列的抗原多肽蛋白的表达性能。
[0062] 实施例21 编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 2所示核苷酸序列的ires元件,具有seq id 47所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂(seq id 59),以及可用
于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,220rpm活化3小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(245.7纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(220.6纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在36.9摄氏度孵育2小时,然后加入dnase i在37摄氏度消化20分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(402.8纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在55摄氏度加热25分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(212.4纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在37摄氏度加热20 分钟,随后在85摄氏度加热10秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为37次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于37摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于36.9摄氏度,5%co2培养箱中培养。待细胞达到90%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为120.3纳克每毫升,167.9纳克每毫升,272.1纳克每毫升,175.9纳克每毫升,108.4纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 47所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分
别为12.2纳克每毫升,19.4纳克每毫升,39.2纳克每毫升,21.3纳克每毫升,10.7纳克每毫升)具有更出色的seq id 47所示氨基酸序列的抗原多肽蛋白的表达性能。
[0063]
实施例22编码抗原多肽环状mrna的制备及在293t细胞的蛋白表达验证第一步,构建含有启动子(seq id 50),5
´
同源臂(seq id 51),3
´
内含子(seq id 52),第二外显子(seq id 53),5
´
间隔区(seq id 54),具有seq id 3所示核苷酸序列的ires元件,具有seq id 49所示氨基酸序列的肿瘤抗原多肽编码区,3
´
间隔区 (seq id 56),第一外显子 (seq id 57),5
´
内含子 (seq id 58),3
´
同源臂 (seq id 59),以及可用于质粒线性化的酶切位点xbai (seq id 60)的表达抗原多肽目的基因,将所得基因片段连接到puc57载体;第二步,将合成的穿刺菌在36.9摄氏度,223rpm活化3.4小时后,取活化菌液在36.9摄氏度,220rpm培养过夜。然后,抽提质粒,测定od值和质粒提取浓度(279.2纳克每微升);第三步,将10微克质粒,5微升酶(1000units), 50微升10
×
cutsmart buffer和若干体积无酶水混合合成的总体积为500微升的混合液在37.1摄氏度酶切过夜,然后回收酶切产物,进一步进行od值检测并同时用1%琼脂糖凝胶电泳对酶切产物进行鉴定。将达到纯化要求的线性质粒模板用于下一步的体外转录;测定质粒酶切线性化后dna浓度(252.3纳克每微升);第四步,将1微克质粒模板,2微升10
×
reaction buffer,2微升浓度为20毫摩尔每升的atp,2微升浓度为20毫摩尔每升的ctp,2微升浓度为20毫摩尔每升的utp,2微升浓度为20毫摩尔每升的gtp, 2微升t7 rna polymerase mix,及若干体积无核酸酶水构成的总体积为20微升的混合液在37.1摄氏度孵育2.0小时,然后加入dnase i在37摄氏度消化23分钟。将所获得的混合物采用用硅膜离心柱法进行进一步纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对rna大小进行识别;测定mrna转录纯化后rna浓度(527.8纳克每微升);第五步,将25微克mrna溶液中,50微升20毫摩尔每升gtp溶液和若干体积由50毫摩尔每升的tris-hcl,10毫摩尔每升的mgcl2及1毫摩尔每升的dtt混合合成的缓冲液进行混合,然后把所获得的总体积为500微升的混合溶液在56.5摄氏度加热24分钟。将所获得的混合物采用使用硅膜离心柱法进行纯化。对纯化产物进行od值测定,并同时通过1%变性琼脂糖凝胶电泳对环状rna大小进行鉴定;测定mrna环化并纯化后rna浓度(256.5纳克每微升);第六步,将1微克环化或未环化的mrna溶液,4微升rt prmier, 1微升 primerscript rt enzye mix i和若干体积无核酸酶水混合合成的总体积20微升的混合液在36.9摄氏度加热21 分钟,随后在85摄氏度加热11秒,最后在4摄氏度保存;第七步,将第六步所获得的1微升逆转录溶液与2微升10
×
buffer,1.6微升dntp,1微升10微摩尔每升primer-f,1微升10微摩尔每升primer-r,0.5微升taq酶,12.9微升无核酸酶水进行进一步混合。然后将混合液依次按在95摄氏度放置1分钟,95摄氏度放置30秒,60摄氏度放置30秒,72摄氏度放置30秒,72摄氏度放置7分钟(循环次数为38次); 4摄氏度放置保存的pcr程序进行扩增。对扩增产物进行核酸电泳,切胶回收,并进一步纯化,并对纯化产物进行纯化和测序。测序结果显示产物包含连接后的第二外显子和第一外显子序列。这表明已成功制备环状 rna;
第八步,先将293t接种于含有10%胎牛血清,1%双抗的dmem高糖培养基中,于36.9摄氏度,5%co2培养箱中培养。与此同时,将细胞每隔2天进行传代培养。转染时,先将293t细胞以1
×
105个/孔接种于24孔板中,并于37摄氏度,5%co2培养箱中培养。待细胞达到88%融合度后,使用转染试剂将所制备的mrna以500纳克/孔量转染293t细胞;第九步,通过elisa试剂盒检测所制备的环状mrna在293t表达第一天到第五天所得蛋白的量分别为33.5纳克每毫升,57.3纳克每毫升,82.5纳克每毫升,65.9纳克每毫升,30.4纳克每毫升,验证了本发明所提供的环状mrna表达具有seq id 49所示氨基酸序列的抗原多肽蛋白的高效和持久性,及比相应线性mrna(第一天到第五天所得蛋白的量分别为6.1纳克每毫升,12.3纳克每毫升,19.5纳克每毫升,11.5纳克每毫升,5.5纳克每毫升)具有更出色的seq id 49所示氨基酸序列的抗原多肽蛋白的表达性能。
[0064]
应当理解,以上所述的仅是本发明的一些实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明的创造构思的前提下,还可以做出其它变状和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1