一种伪模板分子印迹聚合物及其应用的制作方法
一种伪模板分子印迹聚合物及其应用
1.本技术为申请日为2020年07月07日,申请号为202010645743.x,发明创造名称为“一种伪模板分子印迹聚合物及其应用”的分案申请。
技术领域
2.本发明涉及化合物制备技术领域,尤其涉及一种伪模板分子印迹聚合物及其应用。
背景技术:
3.自1971年紫杉醇首次从太平洋紫杉树的树皮中分离出来后,紫杉醇的主要来源仍然是红豆杉树皮。由于紫杉醇对癌症特别是乳腺癌和卵巢癌惊人的治疗作用,独特的抗癌机制,新颖的结构和有限的自然资源,紫杉醇的分离和纯化仍然是重要的课题。
4.目前已经开发了几种分离纯化紫杉醇的方法,包括从红豆杉树皮中直接提取,使用10-去乙酰基巴卡亭ⅲ为起始原料合成紫杉醇,以及植物细胞生物合成法等。然而,由于紫杉醇是不稳定的,在酸,碱,温度和其他条件的影响下易于异构化和降解。同时,较低的分离效率导致分离纯化成本居高不下。因此,迫切需要建立一种快速,高效,经济,环保的紫杉醇分离纯化方法。
技术实现要素:
5.有鉴于此,本发明要解决的技术问题在于提供一种伪模板分子印迹聚合物及其应用,可以从紫杉醇粗提物中有效的富集、分离、纯化紫杉醇,或作为紫杉醇的载体,用于在胃肠道中释放紫杉醇。
6.本发明提供了一种伪模板分子印迹聚合物,以10-去乙酰基巴卡亭ⅲ或紫杉醇侧链为模板,低共熔溶剂为功能单体,偶氮二异丁腈(aibn)为引发剂,乙二醇二甲基丙烯酸酯(egdma)为交联剂,进行聚合反应,然后去除模板分子,制备得到。
7.分别记为10-dab-mips和sc-mips。
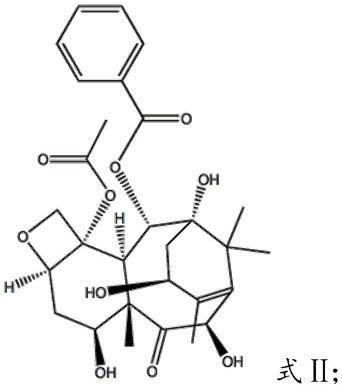
8.其中,10-去乙酰基巴卡亭ⅲ结构如式ⅱ所示:
[0009][0010]
紫杉醇侧链结构如式ⅰ所示:
[0011][0012]
本发明优选的,所述低共熔溶剂包括咖啡酸,氯化胆碱和甲酸。
[0013]
优选的,所述咖啡酸,氯化胆碱和甲酸的摩尔比为1:(1~6):(3~6)。
[0014]
在本发明的一些具体实施例中,所述咖啡酸,氯化胆碱和甲酸的摩尔比为1:5:5,1:6:6,1:6:3,优选1:6:3。在该比例下,制备的伪模板分子印迹聚合物对紫杉醇具有较高的吸附性能。
[0015]
本发明优选的,所述模板、低共熔溶剂,偶氮二异丁腈,乙二醇二甲基丙烯酸酯的用量比为(20-22)mg:(0.5-3)ml:(18-21)mg:(0.5-3)ml。
[0016]
进一步优选为21.7mg:1.0ml:20mg:1.0ml。
[0017]
本发明优选的,所述聚合反应的溶剂为甲醇和三氯甲烷的混合溶剂。
[0018]
本发明优选的,首先采用甲醇使模板分子溶解,然后加入三氯甲烷进行聚合反应。
[0019]
所述甲醇和三氯甲烷的体积比为(1~3):(3~5)。
[0020]
在本发明的一些具体实施例中,所述甲醇和三氯甲烷的体积比为1:2、1:3、1:5或3:5;优选1:5。在该比例下,制备的伪模板分子印迹聚合物对紫杉醇具有较高的吸附性能。
[0021]
本发明提供了伪模板分子印迹聚合物的制备方法,包括以下步骤:
[0022]
s1)模板分子和低共熔溶剂在甲醇和三氯甲烷的混合溶剂中进行预聚合;所述模板分子为10-去乙酰基巴卡亭ⅲ或紫杉醇侧链;
[0023]
s2)将预聚合后的物料与偶氮二异丁腈和乙二醇二甲基丙烯酸酯混合,进行聚合反应;
[0024]
s3)用甲醇-乙酸洗涤聚合反应后得到的固体,去除模板分子,得到伪模板分子印迹聚合物。
[0025]
所述低共熔溶剂优选由咖啡酸,氯化胆碱和甲酸反应得到。
[0026]
在本发明的一些具体实施例中,所述咖啡酸,氯化胆碱和甲酸的摩尔比为1:5:5,
1:6:6,1:6:3,优选1:6:3。
[0027]
所述预聚合的温度优选为室温。
[0028]
所述步骤s2)中,聚合反应优选为50-70℃,进一步优选60℃,聚合5~8h,然后60-80℃,进一步优选70℃,聚合15~20h。
[0029]
本发明优选的,聚合反应后,将聚合产物研磨过筛,并在甲醇中反复清洗,以获得小颗粒。
[0030]
所述步骤s3)中,甲醇-乙酸的体积比优选为8-9:1-15。
[0031]
实验结果表明,本发明制备的低共熔溶剂作为功能单体的分子印迹聚合物为均匀分散的微球,具有良好的热稳定性,对紫杉醇具有优良的吸附性和选择性,可作为紫杉醇载体,在人体肠道中具有良好的可释放性。
[0032]
本发明提供了上述伪模板分子印迹聚合物或上述制备方法制备的伪模板分子印迹聚合物在富集、分离、纯化紫杉醇,或作为紫杉醇载体的应用。
[0033]
与现有技术相比,本发明提供了一种伪模板分子印迹聚合物,以10-去乙酰基巴卡亭ⅲ或紫杉醇侧链为模板,低共熔溶剂为功能单体,偶氮二异丁腈为引发剂,乙二醇二甲基丙烯酸酯为交联剂,进行聚合反应,然后去除模板分子,制备得到。本发明制备的伪模板分子印迹聚合物对紫杉醇显示出较高的吸附量(分别为23.58mg/g和21.64mg/g)和高选择性,可用于富集紫杉醇。并且可以作为载体,在人体肠道或胃液中释放紫杉醇,洗脱率约45%。结果表明,上述伪模板分子印迹聚合物作为高效富集紫杉醇的填料及复杂生物环境中的药物输送系统具有潜在的应用价值和前景。
附图说明
[0034]
图1为10-dab-mips和sc-mips的扫描电镜图;
[0035]
图2为10-dab-mips和sc-mips的红外光谱图和热重曲线图;
[0036]
图3为des用量以及egdma用量对10-dab-mips和sc-mips吸附量影响的柱状图;
[0037]
图4为10-dab-mips和sc-mips的动态吸附曲线和静态吸附曲线;
[0038]
图5为10-dab-mips和sc-mips在人工胃液、人工肠液和病人粪便中的释放柱状图。
具体实施方式
[0039]
为了进一步说明本发明,下面结合实施例对本发明提供的伪模板分子印迹聚合物及其应用进行详细描述。
[0040]
以下新鲜的粪便样本由重庆肿瘤医院提供。
[0041]
实施例1制备伪模板分子印迹聚合物
[0042]
1-1)制备低共熔溶剂
[0043]
选择不同摩尔比的咖啡酸,氯化胆碱,甲酸(摩尔比分别为1:1:5,1:5:5,1:6:6和1:6:3,如表1所示),加入到100ml的圆底烧瓶中,在90℃的油浴锅中不断搅拌直至形成均匀的黑褐色液体,冷却至室温,分别记为样品des-1、样品des-2、样品des-3和样品des-4,选择最佳状态的(液体)低共熔溶剂,用于分子印迹聚合物的制备。
[0044]
表1咖啡酸,氯化胆碱,甲酸摩尔比及编号
[0045][0046]
结果显示des-1(1:1:5)在90℃下不能形成均匀的液体。des-2(1:5:5),des-3(1:6:6)和des-4(1:6:3)在室温下均为透明液体。
[0047]
1-2)制备伪模板分子印迹聚合物10-dab-mips
[0048]
在圆底烧瓶中加入27.3mg的10-dab,然后加入1ml甲醇使其溶解,然后添加5ml三氯甲烷溶液。将上述混合溶液超声处理10min。然后在圆底烧瓶中加入1.0ml的低共熔溶剂des-4。将圆底烧瓶在恒温振荡器中以105rpm,25℃进行预聚合12h。预聚合后,加入20mg aibn,1.0ml egdma,充氮气,在氮气保护下在60℃下反应6h,70℃下反应18h。反应结束后,将合成的伪模板分子印迹聚合物10-dab-mips粉碎并研磨,然后在索氏提取器中用甲醇/冰醋酸(v:v,9/1)混合溶液洗脱24h,以除去模板分子。洗脱模板后,将伪模板分子印迹聚合物用去离子水洗涤3次,在真空干燥箱中45℃干燥12h。
[0049]
1-3)制备伪模板分子印迹聚合物sc-mips
[0050]
在圆底烧瓶中加入21.7mg的紫杉醇侧链,然后加入1ml甲醇使其溶解,然后添加5ml三氯甲烷溶液。将上述混合溶液超声处理10min。然后在圆底烧瓶中加入1.0ml的低共熔溶剂des-4。将圆底烧瓶在恒温振荡器中以105rpm,25℃进行预聚合12h。预聚合后,加入20mg aibn,1.0ml egdma,充氮气,在氮气保护下在60℃下反应6h,70℃下反应18h。反应结束后,将合成的伪模板分子印迹聚合物sc-mips粉碎并研磨,然后在索氏提取器中用甲醇/冰醋酸(v:v,9/1)混合溶液洗脱24h,以除去模板分子。洗脱模板后,将伪模板分子印迹聚合物用去离子水洗涤3次,在真空干燥箱中45℃干燥12h。
[0051]
实施例2伪模板分子印迹聚合物的表征
[0052]
(1)形态表征
[0053]
采用sem对制备的10-dab-mips和sc-mips的表面形态进行表征,结果如图1所示。
[0054]
图1中,图a和图b为10-dab-mips的扫描电镜图,图c和图d为sc-mips的扫描电镜图。
[0055]
图1中图a~d均显示出相对球形的结构和较好的分散性,证明伪模板分子印迹聚合物的聚合过程较为成功。
[0056]
(2)红外光谱分析
[0057]
采用红外光谱分析伪模板分子印迹聚合物材料的官能团。红外光谱具有两个区域:官能团区域(4000-1-333cm-1
)和指纹区域(1333-1-400cm-1
)。10-dab-mips和sc-mips的红外光谱如图2所示。
[0058]
其中,图2中图a为10-dab-mips和sc-mips的红外光谱图。
[0059]
可以看出,对于10-dab-mips和sc-mips都观察到了几个特征峰,其中包括一个从3200-1-3500cm-1
的相似的宽峰,它对应于o-h拉伸振动。在2971cm-1
处包含一个分裂双峰,对应于c-h的对称拉伸,表明低共熔溶剂中双键的成功参加反应。1800cm-1
处的一个强峰,对应于聚合物中c=o的强拉伸振动,1405cm-1
处的一个峰对应于苯环的振动。基于以上结果,证实了低共熔溶剂参与了伪模板分子印迹聚合物的合成。
[0060]
(3)热重分析
[0061]
采用热重分析表征10-dab-mips和sc-mips的热稳定性。实验条件:在空气氛围中以10℃/min的加热速率进行热重分析实验。图2中图b显示出了热重分析曲线。
[0062]
由图2中图b可以看出,在200℃以下时,伪模板分子印迹聚合物约有5%的重量损失,这主要是由于样品水分的消除。当温度超过300℃时,由于热解分解而导致大的重量损失约80%。实验结果证明10-dab-mips和sc-mips在300℃以下时具有良好的稳定性。
[0063]
实施例3反应溶剂的影响
[0064]
按照实施例1相同的步骤制备sc-mips,反应溶剂如表2所示。根据对紫杉醇的吸附量,如表2所示,mip-4(15.18mg/g)对紫杉醇的吸附能力为最佳。因此甲醇:三氯甲烷=1:5为最佳反应溶剂。
[0065]
表2溶剂体系对合成分子印迹聚合物的影响
[0066][0067]
实施例4功能单体和交联剂体积的影响
[0068]
按照实施例1相同的步骤分别制备10-dab-mips和sc-mips,功能单体的用量分别为0.2、0.5、1.0、2.0、3.0ml,测试产物吸附紫杉醇的量,结果如图3中图a所示,图a为des用量对合成10-dab-mips和sc-mips的影响柱形图。可以看出,当功能单体的体积低于1.0ml时,吸附量q值与功能单体的含量呈正相关。当功能单体的体积大于1.0ml时,吸附量q值减小而不是增加。因此功能单体的最佳剂量为1.0ml。
[0069]
按照实施例1相同的步骤分别制备10-dab-mips和sc-mips,egdma的用量分别为0.2、0.5、1.0、2.0、3.0ml,测试产物吸附紫杉醇的量,结果如图3中图b所示,图b为egdma用量对合成10-dab-mips和sc-mips的影响柱形图。可以看出,当egdma的体积低于1.0ml时,吸附量q值与功能单体的含量呈正相关。当egdma的体积大于1.0ml时,吸附量q值减小而不是增加。因此egdma的最佳剂量为1.0ml。
[0070]
比较例1
[0071]
不加入模板分子,其余步骤同实施例1,制备非分子印迹的聚合物,记为nips。
[0072]
实施例5吸附性能考察
[0073]
为了研究10-dab-mips和sc-mips的吸附性能,进行了动态吸附,静态吸附和选择
性吸附实验。
[0074]
5-1)动态吸附试验
[0075]
取10ml离心管,分别加入10.0mg伪模板分子印迹聚合物材料10-dab-mips和sc-mips,然后加入5ml紫杉醇标准溶液(100ug/ml)。吸附过程在105rpm和25℃下进行,分别吸附5、20、40、60、80、120和180min。吸附结束后,通过hplc检测上清液中紫杉醇的浓度。每组实验平行重复三次。伪模板分子印迹聚合物的吸附量通过以下公式计算:
[0076]
q=(c
0-c
t
)v/m
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4.1)
[0077]
其中q(mg/g)为吸附量。c0(μg/ml)和c
t
(μg/ml)是紫杉醇的初始浓度和时间为t时的浓度。v(ml)和m(mg)分别是溶液的体积和伪模板分子印迹聚合物材料的质量。
[0078]
对非分子印迹聚合物以相同的实验方法进行动态吸附性能考察。
[0079]
结果如图4所示。
[0080]
图4中图a为10-dab-mips和sc-mips的动态吸附曲线图,显示了10-dab-mips和sc-mips在室温下的动态吸附数据。可以看出,在开始的60min内,10-dab-mips和sc-mips的吸附量迅速增加,并在180min时达到吸附平衡。非分子印迹聚合物材料的趋势与分子印迹聚合物材料相似,但它的吸附量低于伪模板分子印迹聚合物。这是由于在聚合过程中,洗脱模板后,聚合物骨架中保留了一些与模板分子结构、尺寸相似的结合位点,因此吸附能力更高。而对于非分子印迹聚合物,由于缺乏这样的识别空腔,因此吸附能力较低。
[0081]
为了进一步研究动态吸附过程,采用一级动力学模型和二级动力学模型模拟分子印迹聚合物的动态吸附过程。其公式如下:
[0082]
一级动力学:ln(q
e-q
t
)=lnq
e-k1t
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4.3)
[0083]
二级动力学:t/q
t
=t/qe+1/q
e2
k2ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4.4)
[0084]
其中,k1(min-1
)和k2(g/(mg
×
min))分别表示一级动力学反应速率常数和二级动力学反应速率常数,t代表吸附时间。qe和q
t
代表最大吸附量和吸附时间为t时的吸附量。拟合数据结果列于表3,二级动力学拟合曲线如图4中图b所示。二级动力学方程(r
210-dab-mips
=0.992;r
2sc-mips
=0.953)的r2优于一级动力学方程(r
210-dab-mips
=0.894;r
2sc-mips
=0.698)。此外,二级动力学方程拟合计算的qe值与实验数值相似,也表明吸附动力学数据与二级动力学模型吻合良好。根据拟二级动力学方程机理,化学相互作用可能是伪模板分子印迹聚合物吸附过程中的限速步骤。
[0085]
表3 10-dab-mips和sc-mips的一级动力学和二级动力学计算参数
[0086][0087]
5-2)静态吸附实验
[0088]
将10.0mg 10-dab-mips和sc-mips分别分散在5.0ml一系列不同浓度的紫杉醇标准溶液中,其浓度范围为10-300μg/ml。将这些混合物在25℃下振荡12h。吸附反应结束后,
通过hplc检测上清液中紫杉醇的浓度。10-dab-mips和sc-mips对紫杉醇的吸附容量根据方程(4.1)计算。
[0089]
对非分子印迹聚合物以相同的实验方法进行静态吸附性能考察。
[0090]
实验结果如图4中图c所示,如c是10-dab-mips、sc-mips和nips的静态吸附曲线图。
[0091]
可以看出,10-dab-mips和sc-mips对紫杉醇的吸附量随着紫杉醇的初始浓度的增加而增加,直至达到平衡水平。10-dab-mips和sc-mips的吸附容量分别在紫杉醇浓度为200μg/ml和300μg/ml时达到最大值,其最大吸附量分别为15.48mg/g和19.23mg/g。非分子印迹聚合物与10-dab-mips和sc-mips相比,对紫杉醇的吸附能力较低,这表明伪模板分子印迹聚合物作为吸附材料具有丰富的识别位点用于富集紫杉醇。
[0092]
采用langmuir和freundlich模型进一步描述10-dab-mips和sc-mips对紫杉醇的静态吸附过程。
[0093]
langmuir和freundlich方程如下:
[0094]
langmuir model:1/qe=1/(k
lce
qm)+1/qmꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4.5)
[0095]
freundlich model:logqe=mlogce+logα
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4.6)
[0096]
其中k
l
是langmuir常数,m和α是freundlich常数。qm和qe代表最大吸附量和在浓度为ce时的吸附量。模拟数据列于表4,拟合曲线如图4中图d所示,结果表明langmuir方程(r2》0.97)可以更好地拟合静态吸附数据。可以推测,伪模板分子印迹聚合物材料对紫杉醇吸附过程可能是单层吸附。
[0097]
表4 10-dab-mips和sc-mips的langmuir和freundlich的计算参数
[0098][0099]
5-3)选择性考察
[0100]
选择多西他赛,10-去乙酰基巴卡亭ⅲ和葡萄糖(因为红豆杉中含有大量葡萄糖)作为干扰分子,进行特异性吸附实验,其结构分别如下所示,其中,a为紫杉醇的结构式,b为10-去乙酰基巴卡亭ⅲ的结构式,c为紫杉醇侧链的结构式。选用60%的甲醇溶液配制100μg/ml的多西他赛,10-去乙酰基巴卡亭ⅲ和葡萄糖混合溶液。取10ml离心管,准确称取10.0mg的伪模板分子印迹聚合物材料加入其中,然后加入5ml上述混合溶液,在恒温振荡器中室温振摇2h。振摇结束后,取上清液,然后通过hplc检测上清液中紫杉醇的浓度。10-dab-mips和sc-mips对紫杉醇的吸附量qe(mg/g)的计算方法同方程式(4.1)。
[0101][0102]
实验结果如图4中图e所示,图e是10-dab-mips、sc-mips和nips的选择性吸附柱状图。
[0103]
可以看出,伪模板分子印迹聚合物对紫杉醇的吸附能力远高于葡萄糖,略高于另外两个干扰物质(10-去乙酰巴卡亭ⅲ和多西他赛)。这很可能是由于10-去乙酰巴卡亭ⅲ以及多西他赛和紫杉醇的结构相似。可以预测这种伪模板分子印迹聚合物可用于同时富集红豆杉中结构相似的分子,并且不受其他物质的影响。
[0104]
采用印迹因子(if)来评估10-dab-mips和sc-mips的特异性吸附能力。
[0105]
if=q
mips
/q
nips
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(4.7)
[0106]
计算出的印迹因子列于表5。10-dab-mips对紫杉醇,10-去乙酰基巴卡亭ⅲ,多西他赛和葡萄糖,其印迹因子分别计算为2.38、2.85、1.41和1.84。sc-mips对紫杉醇,10-去乙酰基巴卡亭ⅲ,多西他赛和葡萄糖的印迹因子分别计算为2.72、2.57、1.34和1.02。可以看出10-dab-mips和sc-mips具有较高的选择性和吸附能力,可用于同时从复杂基质中分离紫杉醇,10-去乙酰基巴卡亭ⅲ和多西他赛。
[0107]
表5 10-dab-mips,sc-mips和nips的选择性参数
[0108][0109]
实施例6应用
[0110]
6-1)考察伪模板分子印迹聚合物在人工胃液和人工肠液中的洗脱
[0111]
人工胃液的配制配置:准确量取1.64ml稀盐酸,然后加入约80ml的去离子水,并准确称取1g的胃蛋白酶溶于上述混合溶液中,用去离子水定容至100ml,存放于4℃冰箱备用。人工肠液的配制:称取磷酸二氢钾6.8g,然后加入500ml去离子水使溶解,溶解后用0.1mol/l的氢氧化钠溶液调节ph值至6.8;另取胰蛋白酶10g,加适量去离子水使溶解,溶解后与上述溶液混合,然后加去离子水稀释至1000ml,即得。
[0112]
首先制备饱和的伪模板分子印迹聚合物材料:称取10mg的10-dab-mips和sc-mips分别添加到含有5ml,100μg/ml的紫杉醇溶液中,并在恒温振荡器中以105rpm,25℃吸附12h。吸附结束后,通过离心除去上清液,并用去离子水洗涤饱和的10-dab-mips和sc-mips洗涤3次。将5ml人工胃液和人工肠液分别添加到上述饱和的分子印迹聚合物中,并在105rpm,37℃下振动5、10、20、30和60min进行洗脱。当洗脱过程完成后,将试管中的人造胃液和人造肠液在45℃下干燥,然后用5ml甲醇添加到试管中以重新溶解紫杉醇。实验结束后,通过hplc测定上清液,母液和洗脱液中紫杉醇的浓度,并根据公式(4.2)计算的10-dab-mip和sc-mips的洗脱百分比。本次实验设置了3个平行组。
[0113]
释放百分比=c
洗脱
×v洗脱
/(c
0-ce)*v
吸附
×
100%(4.2)
[0114]
其中,c
洗脱
表示洗脱液中紫杉醇的浓度,c0吸附前紫杉醇的浓度,ce表示吸附后紫杉醇的浓度,v
洗脱
表示洗脱液的体积,v吸附表示吸附时的体积。
[0115]
实验结果如图5所示,其中,图5中图a为10-dab-mips和sc-mips在人工胃液中释放紫杉醇柱状图;图b为10-dab-mips和sc-mips在人工肠液中释放紫杉醇柱状图。
[0116]
结果表明,在人工胃液中,在5min内从10-dab-mips中释放出约45%的紫杉醇。同样,在人工肠液中,大约52%的紫杉醇在10min内从sc-mips中被释放出来。可以看出,10-dab-mips和sc-mips可以在人工胃液中释放出来。在人工胃液中10-dab-mips的释放速度较快,而在人工肠液中sc-mips的释放速率较快。
[0117]
6-2)考察伪模板分子印迹聚合物在新鲜粪便中的洗脱
[0118]
病人的粪便由重庆市肿瘤医院(中国重庆)提供。首先将4g粪便加入20ml生理盐水溶液中并涡旋3min。随后,将粪便悬浮液以10000g离心20min,然后收集上清液。取8支10ml离心管,加入20mg饱和的10-dab-mips和sc-mips聚合物材料(吸附紫杉醇后),然后加入5ml粪便上清液,密封。将离心管在恒温振荡器上以105rpm的速度,37℃摇动2h。振摇结束后,离心,取上清液,于45℃烘干,然后用甲醇复溶,随后用hplc分析其中紫杉醇的浓度,并按照(4.2)式计算伪模板分子印迹聚合物释放紫杉醇的百分比。
[0119]
实验结果如图5所示,其中,图5中图c为10-dab-mips和sc-mips在病人粪便中释放紫杉醇柱状图。
[0120]
结果表明,在新鲜的粪便样品中,从10-dab-mips和sc-mips中释放出约30%-35%的紫杉醇,在粪便样品中成功检测到紫杉醇且有着较高的释放率。
[0121]
以上释放实验结果表明,10-dab-mips和sc-mips作为紫杉醇载体,可以在不同条件下成功释放紫杉醇。本发明制备的伪模板分子印迹的聚合物将来可以用作药物载体。
[0122]
以上液相分析条件如下:
[0123]
选用安捷伦1260型高效液相色谱仪,c
18
色谱柱(250
×
4.6mm,5μm);柱温30℃;流速0.8ml/min;进样量10μl;流动相70%的乙腈(a)-纯水(b),等度洗脱,检测波长为227nm。
[0124]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1