一类1,3-氮杂硅烷化合物的合成方法及其应用
1.本发明涉及一类1,3-氮杂硅烷化合物的合成方法及其应用,属于硅氮化合物化学合成技术领域。
背景技术:
2.1,3-氮杂硅烷化合物是一类简单、高效的构建硅氮杂环的合成子。2000年,voronkov课题组报道了含有烯基或烯丙基硅的1,3-氮杂硅烷化合物在hg(oac)2催化下通过分子内氮原子亲核进攻硅基β位不饱和碳原子发生环化反应生成硅代吡咯烷(图1);2014年,maddauno课题组报道了邻位连接有1,3-氮杂硅烷的溴苯通过锂溴交换生成的超价硅环经sila-matteson重排生成两种形式的苯并硅氮六元杂环(图2);2018年,hartwig课题组报道了含有脂肪胺基的1,3-氮杂硅烷在过渡金属铱的催化下选择性对胺基β位c(sp3)-h键的硅烷化反应(图3);2021年,song课题组报道了碱性条件下1,3-氮杂硅烷化合物原位生成氮杂环丁烷后经过pd(pph3)2cl2催化与端炔类化合物发生[4+2]扩环反应生成六元硅氮杂环(其中,具体涉及脱去si处α位的x基团生成四元环的结构,图4)。
[0003]
目前,基于1,3-氮杂硅烷化合物构建的硅氮杂环仅限于五元、六元硅氮杂环及苯并六元硅氮杂环化合物,未有以1,3-氮杂硅烷化合物为合成子构建苯并硅氮五元杂环的报道。
[0004]
吲哚啉为一种苯并氮杂五元环化合物,其广泛存在于天然产物及活性小分子化合物母体结构中(如图5)。碳原子与硅原子同属iv族,具有相似的物理及化学性质,是理想的生物电子等排体,通过生物电子等排体策略进行c/si转换生成的苯并硅氮五元杂环-硅代吲哚啉,其在药物研究中具有重要的研究价值,这可为药物研发前期的先导化合物结构改造提供优化的思路和方向,比如:2019年,driver课题组报道了dmgs-rar-5
·
ots可作为烟酰胺磷酸核糖转移酶抑制剂(ic
50 = 10.3 nm)(图6)。对于硅代吲哚啉的合成,目前,仅有两种合成硅代吲哚啉的方法报道。2017年,huang课题组通过ru催化2-二甲氨基取代的苯基硅氢化合物,经过分子内的 c(sp3)-h脱氢硅烷化反应得到硅代吲哚啉(图7);2019年,driver课题组报道了(2-叠氮苯基)三甲基硅烷在rh2(esp)2催化下与二碳酸二叔丁酯反应得到硅代吲哚啉(图8)。但这两种方法皆是通过预制含有硅氮取代的反应底物发生分子内环化反应得到目标产物,且使用有害的过渡金属催化剂,从而会导致环境污染等问题;此外,由于需要在底物上预制含有硅氮的官能团,导致底物合成难度大,适用范围较为局限。
[0005]
因此,急需开发一类1,3-氮杂硅烷化合物的合成方法,并以1,3-氮杂硅烷化合物为合成子设计一种简单高效的构建硅代吲哚啉的方法。
技术实现要素:
[0006]
本发明针对现有技术的不足,而提出了一类1,3-氮杂硅烷化合物的合成方法及其应用。在本技术方案中,本发明人提出新的一类前体化合物-1,3-氮杂硅烷化合物的合成方法,并以此为起始原料,用于制备硅代吲哚啉或者其他药物组合物,保证后续制备方法易操
作、涉及试剂危险性低、反应条件不苛刻等,进而保证1,3-氮杂硅烷化合物为反应底物时的适用性强,同时,实现硅代吲哚啉的合成,为新药研发等提供可参考的前体化合物选择。
[0007]
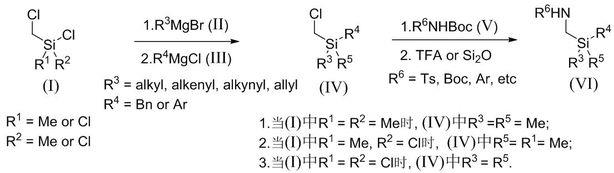
为了实现上述技术目的,提出如下的技术方案:本技术方案提出:一类1,3-氮杂硅烷化合物的合成方法,包括如下步骤:a. 将如式(ⅰ)所示化合物与如式(ⅱ)所示的化合物和/或如式(ⅲ)所示化合物依次进行亲核取代反应,即得如式(ⅳ)所示化合物;b.在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:其中,r1为甲基或氯基,r2为甲基或氯基,r3为烷基、烯基、炔基或芳基,r4为苄基或芳基,r5为烷基、烯基、炔基或芳基,r6为磺酰基、酰基、烷基或芳基。
[0008]
当式(ⅰ)所示反应底物不同时(即r1和r2不同),涉及到1,3-氮杂硅烷化合物的合成路线稍有差异,比如:1)r1= r2,且同为氯基;r3= r5,且同为烷基、烯基、炔基或芳基;r4为苄基或芳基;r6为磺酰基、酰基、烷基或芳基;x1:将如式(ⅰ)所示化合物与如式(ⅱ)所示的化合物和如式(ⅲ)所示的化合物依次进行亲核取代反应,即得如式(ⅳ)所示化合物;x2:在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:;2)r1= r2,且同为甲基;r3= r5,且同为甲基;r4为苄基或芳基;r6为磺酰基、酰基、烷基或芳基;y1:将如式(ⅰ)所示化合物与如式(ⅲ)所示的化合物进行亲核取代反应,即得如式(ⅳ)所示化合物;y2:在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:
;3)r1为甲基;r2为氯基;r1= r5,且同为甲基;r3为烷基、烯基、炔基或芳基;r4为苄基或芳基;r6为磺酰基、酰基、烷基或芳基;z1:将如式(ⅰ)所示化合物与如式(ⅱ)所示的化合物和如式(ⅲ)所示化合物依次进行亲核取代反应,即得如式(ⅳ)所示化合物;z2:在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:。
[0009]
优选的,所述1,3-氮杂硅烷化合物包括并不限于:
ꢀꢀꢀꢀꢀ
。
[0010]
优选的,在步骤a中,亲核取代反应时,反应溶剂为四氢呋喃,反应温度为0℃~室温;式(ⅱ)所示化合物和式(ⅲ)所示化合物的加入速度均为10 ml/h。
[0011]
优选的,在步骤a中,式(ⅰ)所示化合物、式(ⅱ)所示化合物和式(ⅲ)所示化合物三者物质的量配比为1.0 : 1.0~2.2:1.0~1.2,式(ⅱ)所示化合物和式(ⅲ)所示化合物于反应溶剂中的浓度均为0.3~0.5mol/l。
[0012]
优选的,在步骤b的亲核取代反应中,碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、碳酸铯及氢化钠中的一种或任意两种以上的组合,反应溶剂为n,n-二甲基甲酰胺及二甲基亚砜中的一种或两种的组合,反应温度为60~90℃。
[0013]
优选的,在步骤b的亲核取代反应中,式(ⅳ)所示化合物、式(
ⅴ
)所示化合物及碱三者物质的量配比为1.0 : 1.0~1.2 : 1.0~2.0;式(ⅳ)所示化合物于反应溶剂中的浓度为0.2~0.5mol/l。
[0014]
优选的,在步骤b的脱boc保护基反应中,采用三氟乙酸(tfa)或二氧化硅为脱boc保护基的试剂,其中,反应溶剂为二氯甲烷或甲苯。
[0015]
本技术方案提出:一类采用上述方法合成的1,3-氮杂硅烷化合物,用于制备硅代吲哚啉。
[0016]
优选的,所述制备硅代吲哚啉的方法包括:在碱性条件下,将式(ⅵ)所示化合物与式(ⅶ)所示化合物进行[3+2]环化反应,得到式(
ⅷ
)所示化合物,即硅代吲哚啉;涉及的反应式如下:
其中,r7为烷基、烯基、炔基、芳基或卤原子。
[0017]
优选的,在[3+2]环化反应中,碱为氟化铯、氟化钾、碳酸钾、碳酸铯及氢氧化钾中的一种或两种以上的组合;采用两相催化剂为18-冠醚-6、15-冠醚-5、苯并18-冠醚-6及二苯并18-冠醚-6中的一种或两种以上的组合;涉及反应溶剂为乙酸乙酯、乙醚、乙二醇二甲醚、乙腈、甲苯、四氢呋喃及1,4-二氧六环中的一种或两种以上的组合。
[0018]
优选的,通式(ⅵ)所示化合物与通式(ⅶ)所示化合物之间的物质的量配比为1.0 : 1.0~3.0。
[0019]
本技术方案还提出:一种含采用上述方法合成的硅代吲哚啉的产品,包括硅代吲哚啉化合物或其药学上可用的盐为活性成分,以及药学上可接受的载体、稀释剂和赋形剂组成的药物组合物。
[0020]
优选的,所述产品为镇痛药物、抗肿瘤药物、抗菌药物、抗抑郁药物或血管扩张药物。
[0021]
本技术方案还提出:一种含采用上述方法合成的1,3-氮杂硅烷化合物的产品,包括1,3-氮杂硅烷化合物或其药学上可用的盐为活性成分,以及药学上可接受的载体、稀释剂和赋形剂组成的药物组合物。
[0022]
采用本技术方案,带来的有益技术效果为:1)本发明提出新的一类前体化合物-1,3-氮杂硅烷化合物的合成方法,并以此为起始原料,用于制备硅代吲哚啉或者其他药物组合物,保证后续制备方法易操作、涉及试剂危险性低、反应条件不苛刻等,进而保证1,3-氮杂硅烷化合物为底物时的适用性强,同时,实现硅代吲哚啉的合成,为新药研发等提供可参考的前体化合物选择;2)在本发明中,通过简单易得的原料和常规试剂,在温和条件下以克级规模制备出了1,3-氮杂硅烷化合物。该化合物在碱性条件下,可以与原位生成的苯炔前体在碱性条件下发生反应(分子间环化反应)来构建结构丰富的硅代吲哚啉,该方法可应用于药物结构的后期修饰(药物研发)、新材料制备等;3)在本发明中,在1,3-氮杂硅烷化合物的合成方法中,所涉及的试剂均为商业可得的大宗原料,合成工艺简单,常温即可保存;4)相较于已有两种的硅代吲哚啉合成方法(huang课题组、driver课题组),本发明以1,3-氮杂硅烷化合物为起始原料不需使用有害的过渡金属催化剂,从而降低了环境污染等问题,更适应于工业化大生产。
附图说明
[0023]
图1为现有技术中hg催化的硅代吡咯烷的合成方法;
图2为现有技术中通过sila-matteson重排得到苯并硅氮六元杂环的合成 ;图3为现有技术中ir催化的硅代吡咯烷的合成方法;图4为现有技术中pd催化的硅氮六元环的合成方法;图5为含吲哚啉母核结构的天然产物或活性分子化合物;图6为dgms-rar-5
·
ots的化学结构式;图7为现有技术中ru催化的硅代吲哚啉的合成方法;图8为现有技术中rh催化的硅代吲哚啉的合成方法;图9为本发明中1,3-氮杂硅烷化合物的合成方法;图10为本发明中以1,3-氮杂硅烷化合物为原料进行扩环反应的合成方法;图11为对照例中g和i在完全弗氏佐剂诱导的炎性痛模型中的镇痛作用结果(a);图12为对照例中g和i在完全弗氏佐剂诱导的炎性痛模型中的镇痛作用结果(b)。
具体实施方式
[0024]
下面通过对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0025]
在下述实施例中,涉及的设备包括:sgf 254 薄层层析硅胶板(烟台江友硅胶开发有限公司)、bsa124s 型1/10000天平(赛多利斯科学仪器(北京)有限公司)、2zx-4 型旋片真空泵(上海第二真空泵厂有限公司)、85-2型恒温磁力搅拌器(上海司乐仪器厂)、n-1300 型旋转蒸发仪(上海爱朗仪器有限公司)、cca-1112a 型循环水泵 (上海爱朗仪器有限公司)、psl-1810型低温循环反应器(上海爱朗仪器有限公司)、bruker ac-e400型核磁共振仪、finnigan lcq-deca质谱仪(高分辨质谱)、vector22红外光谱仪(红外光谱);在下述实施例中,涉及的试剂包括:n-(叔丁氧羰基)对甲苯磺酰胺 (上海安耐吉化学有限公司),浓度》98%;氯甲基二甲基氯硅烷 (上海泰坦科技股份有限公司),浓度》98%;(氯甲基)甲基二氯硅烷 (上海泰坦科技股份有限公司),浓度》98%;碳酸钾 (成都科隆化工有限公司),浓度》98%;三氟乙酸 (成都科隆化工有限公司),浓度》98%;2-(三甲基硅基)苯基三氟甲烷磺酸盐 (上海安耐吉化学有限公司),浓度》95%;碳酸铯 (上海泰坦科技股份有限公司),浓度》99.9%;18-冠醚-6 (上海泰坦科技股份有限公司),浓度》99%;超干乙二醇二甲醚 (百灵威科技有限公司),浓度》99.5%;金刚烷甲酰氯 (北京伊诺凯科技有限公司),浓度》97%;
3,3-二甲基吲哚啉 (上海毕得医药科技股份有限公司),浓度》97%。
[0026]
实施例1一类1,3-氮杂硅烷化合物的合成方法,包括如下步骤:a. 将如式(ⅰ)所示化合物与如式(ⅱ)所示的化合物和/或如式(ⅲ)所示化合物依次进行亲核取代反应,即得如式(ⅳ)所示化合物;b.在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:其中,r1为甲基或氯基,r2为甲基或氯基,r3为烷基、烯基、炔基或芳基,r4为苄基或芳基,r5为烷基、烯基、炔基或芳基,r6为磺酰基、酰基、烷基或芳基。
[0027]
当式(ⅰ)所示反应底物不同时(即r1和r2不同),涉及到1,3-氮杂硅烷化合物的合成路线稍有差异,比如:1)r1= r2,且同为氯基;r3= r5,且同为烷基、烯基、炔基或芳基;r4为苄基或芳基;r6为磺酰基、酰基、烷基或芳基;x1:将如式(ⅰ)所示化合物与如式(ⅱ)所示的化合物和如式(ⅲ)所示的化合物依次进行亲核取代反应,即得如式(ⅳ)所示化合物;x2:在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;;2)r1= r2,且同为甲基;r3= r5,且同为甲基;r4为苄基或芳基;r6为磺酰基、酰基、烷基或芳基;y1:将如式(ⅰ)所示化合物与如式(ⅲ)所示的化合物进行亲核取代反应,即得如式(ⅳ)所示化合物;y2:在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:
;3)r1为甲基;r2为氯基;r1= r5,且同为甲基;r3为烷基、烯基、炔基或芳基;r4为苄基或芳基;r6为磺酰基、酰基、烷基或芳基;z1:将如式(ⅰ)所示化合物与如式(ⅱ)所示的化合物和如式(ⅲ)所示化合物依次进行亲核取代反应,即得如式(ⅳ)所示化合物;z2:在碱性条件下,将所得的如式(ⅳ)所示化合物与如式(
ⅴ
)所示化合物进行亲核取代反应;然后脱boc保护基,得到如式(ⅵ)所示化合物,即1,3-氮杂硅烷化合物;涉及的反应式如下:。
[0028]
其中,1,3-氮杂硅烷化合物具体包括:
ꢀꢀꢀꢀꢀ
。
[0029]
在步骤a中,亲核取代反应时,反应溶剂为四氢呋喃,反应温度为0℃~室温;式(ⅱ)所示化合物和式(ⅲ)所示化合物的加入速度均为10 ml/h。
[0030]
在步骤a中,式(ⅰ)所示化合物、式(ⅱ)所示化合物和式(ⅲ)所示化合物三者物质的量配比为1.0 : 1.0~1.2:1.0~1.2,式(ⅱ)所示化合物和式(ⅲ)所示化合物于反应溶剂中的浓度均为0.3~0.5mol/l。
[0031]
在步骤b的亲核取代反应中,碱为碳酸钾、碳酸钠、氢氧化钾、氢氧化钠、碳酸铯及氢化钠中的一种或任意两种以上的组合,反应溶剂为n,n-二甲基甲酰胺及二甲基亚砜中的一种或两种的组合,反应温度为60~90℃。
[0032]
在步骤b的亲核取代反应中,式(ⅳ)所示化合物、式(
ⅴ
)所示化合物及碱三者物质的量配比为1.0 : 1.0~2.2 : 1.0~2.0;式(ⅳ)所示化合物于反应溶剂中的浓度为0.2~0.5mol/l。
[0033]
在步骤b的脱boc保护基反应中,采用三氟乙酸或二氧化硅为脱boc保护基的试剂,其中,反应溶剂为二氯甲烷或甲苯。
[0034]
实施例2一类采用上述方法合成的1,3-氮杂硅烷化合物,用于制备硅代吲哚啉。
[0035]
将1,3-氮杂硅烷化合物用于制备硅代吲哚啉,具体包括:在碱性条件下,将式(ⅵ)所示化合物与式(ⅶ)所示化合物进行[3+2]环化反应,得到式(
ⅷ
)所示化合物,即硅代吲哚啉;涉及的反应式如下:
其中,r7为烷基、烯基、炔基、芳基或卤原子。
[0036]
在[3+2]环化反应中,碱为氟化铯、氟化钾、碳酸钾、碳酸铯及氢氧化钾中的一种或两种以上的组合;采用两相催化剂为18-冠醚-6、15-冠醚-5、苯并18-冠醚-6及二苯并18-冠醚-6中的一种或两种以上的组合;涉及反应溶剂为乙酸乙酯、乙醚、乙二醇二甲醚、乙腈、甲苯、四氢呋喃及1,4-二氧六环中的一种或两种以上的组合。
[0037]
通式(ⅵ)所示化合物与通式(ⅶ)所示化合物之间的物质的量配比为1.0 : 1.0~3.0。
[0038]
实施例3基于实施例2,本实施例还提供:一种含采用上述方法合成的硅代吲哚啉的产品,包括硅代吲哚啉化合物或其药学上可用的盐为活性成分,以及药学上可接受的载体、稀释剂和赋形剂组成的药物组合物。
[0039]
更为具体的,比如:产品为镇痛药物、抗肿瘤药物、抗菌药物、抗抑郁药物或血管扩张药物。
[0040]
实施例4基于实施例1,本实施例还提供:本实施例还一种含1,3-氮杂硅烷化合物的产品,包括1,3-氮杂硅烷化合物或其药学上可用的盐为活性成分,以及药学上可接受的载体、稀释剂和赋形剂组成的药物组合物。
[0041]
实施例5本实施例提供一类1,3-氮杂硅烷化合物的合成方法,包括如下步骤:1) 化合物a的制备,反应式如下:冰浴下,将2.0 g (氯甲基)二甲基氯硅烷加入至20 ml的无水四氢呋喃(溶剂)中,然后缓慢加入2.47 g的苄基氯化镁,滴加完毕后,于室温条件下反应24 h;加20 ml的饱和氯化铵溶液淬灭反应,然后用40 ml的乙酸乙酯萃取三次,饱和食盐水分别洗一次,无水na2so4干燥,减压浓缩除溶剂,再用200-300目的硅胶柱层析,得到1.25 g为无色液体的化合物a,产率45%;2)化合物b的制备,反应式如下:
将2.05 g的化合物tsnhboc和1.3 g的碳酸钾(碱)加入至20 ml的n,n-二甲基甲酰胺(溶剂)中,然后加入1.25 g的化合物a,于90℃条件下反应5 h;加50 ml的水淬灭反应,然后用乙酸乙酯萃取三次,合并有机相,用饱和食盐水洗两次,无水na2so4干燥,减压浓缩除溶剂,再用200-300目的硅胶柱层析,得到1.12 g为无色液体的化合物b,产率41%;3)化合物c的制备,反应式如下:将1.12 g的化合物b加入至15 ml的二氯甲烷(溶剂)中,然后缓慢滴加2.51 g的三氟乙酸(强酸,脱boc保护基),然后于室温条件下反应1 h;冰浴下,缓慢滴加饱和nahco3溶液淬灭反应,分离有机相,水相用二氯甲烷萃取三次后,合并有机相,饱和食盐水洗一次,无水na2so4干燥,减压浓缩除溶剂,再用200-300目的硅胶柱层析,得到540 mg为白色固体的化合物c(即1,3-氮杂硅烷化合物),产率60%。
[0042]
实施例6基于实施例5中的合成方法,还得到一系列的1,3-氮杂硅烷化合物,具体如下:一、1,3-氮杂硅烷化合物1(即实施例5中的化合物c),结构式如下:其中,关于1,3-氮杂硅烷化合物1,白色固体,四步收率为10%,熔点:113.5
–
115.6℃。
[0043]1h nmr (400 mhz, cdcl3) δ 7.71 (d, j = 8.4 hz, 2h), 7.32 (d, j = 8.0 hz, 2h), 7.17 (t, j = 7.2 hz, 2h), 7.09 (t, j = 7.2 hz, 1h), 6.91 (d, j = 7.2 hz, 2h), 4.12 (t, j = 6.0 hz, 1h), 2.44 (s, 3h), 2.28 (d, j = 6.0 hz, 2h), 2.12 (s, 2h), 0.04 (s, 6h).
13
c nmr (100 mhz, cdcl3) δ 143.4, 138.6, 135.3, 129.6, 128.6, 127.9, 127.5, 124.6, 30.8, 24.0, 21.5,
ꢀ‑
4.7.
29
si nmr (119 mhz, chloroform-d) δ
ꢀ‑
1.94.ir (neat) cm-1 3272, 3006, 1321, 1275, 1260, 1160, 849, 750.hrms (esi-tof, m/z) calcd for c
17h23
no2ssi (m+h)
+
: 334.1292, found 334.1292.;二、1,3-氮杂硅烷化合物2,结构式如下:
764, 725.hrms calcd for c
20h27
nosi (m+h)
+
: 326.1935, found 326.1933.。
[0057]
2)通过3,3-二甲基吲哚啉(即化合物h),构建具有镇痛活性的目标化合物具体反应式如下:氩气保护下,将100 mg的化合物h(3,3-二甲基吲哚啉)溶于3 ml 无水二氯甲烷(溶剂)中,随后加入263 mg的二异丙基乙胺和202 mg的金刚烷甲酰氯,室温搅拌2个小时。待tlc监测反应结束后,加入饱和碳酸氢钠溶液淬灭,二氯甲烷萃取三次,饱和食盐水反洗一次,无水硫酸钠干燥,减压浓缩得到的粗产物通过硅胶柱层析分离 (pe:ea = 20:1),得到175 mg的白色固体i,产率83%。熔点:134.8
–
136.5 ℃。
[0058]1h nmr (400 mhz, cdcl3) δ 8.20 (s, 1h), 7.25
ꢀ–ꢀ
6.92 (m, 3h), 4.03 (s, 2h), 2.10 (s, 9h), 1.77 (s, 6h), 1.33 (s, 6h).
13
c nmr (100 mhz, cdcl3) δ 176.0, 143.6, 139.9, 127.4, 123.8, 121.3, 118.6, 63.8, 42.9, 41.0, 38.2, 36.6, 28.4, 27.1.ir (neat) cm-1 3006, 2903, 2989, 2849, 1639, 1597, 1452, 1275, 1260, 764, 751.hrms calcd for c
21h27
no (m+h)
+
: 310.2165, found 310.2163.。
[0059]
3)进行cb1和cb2受体激动剂的ec
50
测试,用以评估化合物g与化合物i对靶点受体的活性及选择性。
[0060]
利用二甲基亚砜将阳性化合物(上述所得的化合物g和化合物i)分别进行梯度稀释,然后用1
×ꢀ
stimulation buffer 将对应化合物稀释至10
×
。
[0061]
cb1和cb2稳转的cho细胞经胰酶消化处理,离心后去除培养基,将细胞重悬于1
×ꢀ
stimulation buffer中,经计数后接种于384-well板中;取1μl稀释好的对应化合物加入至相应实验孔中,放置于37℃孵育。
[0062]
将白喉毒素用1
×ꢀ
stimulation buffer稀释至2.5
×
,取4μl 稀释好的白喉毒素加入至 cb1/cb
2-cho相应实验孔中,离心后放置于37℃孵育;用detection buffer 将 eu-camp 进行50倍稀释,然后取5μl孔加入至相应实验孔中;将ulight
™‑
anti-camp用detection buffer进行150倍稀释,然后取5μl孔加入相应实验孔中;200g离心后放置于室温孵育1小时;孵育完成后,使用biotek多功能读板机检测665nm和615nm 读值,所得结果如下表1所示。
[0063]
由上可知:化合物g与化合物i均对cb2受体具有良好的活性,两者均对cb1受体活性
较低,即两者具有较好的cb2受体选择性;其中,涉及的试剂及耗材如下表2所示。
[0064]
4)小鼠血浆-大脑分布测定,评估化合物g与化合物i在脑内分布和血浆内分布的差异,用以衡量化合物通过血脑屏障进入大脑组织的能力。
[0065]
将12只小鼠随机分组,雌雄各半,约重22~25 g/只。每组为1个取样时点,动物禁食12 h(不禁水),在0.97 mmol/kg的药物剂量下,每只小鼠通过灌胃 1次性给药,于给药40 min后取血。其中,以所得的化合物g和化合物i分别为药物。
[0066]
将小鼠断头,开胸腔进行心脏取血,取完血后立即取出小鼠脑组织,后加0.5 ml生理盐水,用高通量组织破碎仪研磨制成脑匀浆。其中,血浆和脑匀浆均以3500 rpm的速率离心10 min,分离,取100 μl 上清液,加入300 μl乙腈完全沉淀蛋白,涡旋摇匀,用超声波振荡3~5 min,以20000 rpm的速率离心10 min,取适量上清液进行hplc分析,所得结果如下表3所示。
[0067]
由上可知:相较于化合物i(大脑组织/血浆 =0.89),化合物g具有更好的血脑屏障通透性(大脑组织/血浆 = 5.53)。
[0068]
5)建立完全弗氏佐剂诱导的疼痛模型,用以评估化合物g与化合物i的体内镇痛活性。
[0069]
将20μl无菌生理盐水或完全弗氏佐剂(sigma-aldrich, f5881)注射至小鼠左脚掌掌心。造模一天后,将小鼠随机分为:模型组、i
‑ꢀ
0.7 mmol/kg组、 i
‑ꢀ
1.0 mmol/kg 组、 i
‑ꢀ
1.3 mmol/kg 组、g
‑ꢀ
0.7 mmol/kg 组、 g
‑ꢀ
1.0 mmol/kg组、 g
‑ꢀ
1.3 mmol/kg组。将小鼠单独放置于疼痛行为学测试箱中适应30min。使用电子von frey仪(iitc, #2391)测试小
鼠对机械疼痛的反应阈值,所得结果如图11-12所示。
[0070]
其中,von-frey机械痛阈实验中i和g的时-剂量依赖性的镇痛效果,图11中的曲线下面积分析 (auc)(基础值-120 min),每组动物数量n=6。使用单因素或两因素方差分析,及dunnett’s t3或tukey
–
kramer hsd事后检验,分析各组间差异。* p 《 0.05,** p 《 0.01 和 ***p 《 0.001,溶剂组与g-1.3 mmol/kg组相比。&p 《 0.05 和 &&&p 《 0.001,溶剂组与 g
‑ꢀ
1.0 mmol/kg组相比。#p 《 0.05,溶剂组与i-1.3 mmol/kg 和i
‑ꢀ
1.0 mmol/kg组相比。 ##p 《 0.01,溶剂组与i-1.3 mmol/kg组相比。###p 《 0.001,i-1.3 mmol/kg 组与g-1.3 mmol/kg组相比。
[0071]
由上可知:化合物g相较于化合物i具有更好的体内镇痛活性,且化合物g的最大镇痛强度及镇痛时间均优于化合物i,其可取得更好的镇痛收益。
[0072]
讨论例在现有技术(202110297658.3)中,公开:将氮杂环丁硅前体化合物为原料、合成六元硅氮杂环。本讨论例以本技术方案中以实施例7中合成的1,3-氮杂硅烷化合物为原料、合成硅代吲哚啉,以对本发明作进一步的说明。
[0073]
六元硅氮杂环合成方法,具体如下:在手套箱中,将58.4mg的化合物7(即以氮杂环丁硅前体化合物)、7mg的双三苯基磷二氯化钯(催化剂)和12.6mg的碘化锌(稳定剂)加入至10ml的反应管中,然后加入0.5ml的二甲苯、24.6mg的苯乙炔和62mg的1,8-二氮杂环[5,4,0]十一烯-7(碱,攫氢实现分子内关环),密封,在120℃条件下反应10min,冷却至室温,减压浓缩除溶剂,再用200-300目的硅胶柱层析,即得到52mg为白色固体的化合物8(即六元硅氮杂环终产物),产率72%。
[0074]
上述化合物7中,分子结构中的氯亚甲基对应本发明式(ⅵ)所示化合物(即1,3-氮杂硅烷化合物)中的r4基团(苄基或芳基),虽二者化学结构相似,但在后续应用中的反应位点截然不同。化合物7为1,4-氮碳合成子,在碱性条件下该化合物首先断裂c(sp3)-cl键,随后在双三苯基磷二氯化钯催化下与苯乙炔发生分子间[4+2]环化反应,最终制得化合物8,具体如下反应式:;而本发明中,合成硅代吲哚啉的化合物c为1,3-氮硅合成子,其在碱性条件下与原位生成的苯炔中间体通过生成的五价硅中间体断裂c(sp3)-si键发生分子间[3+2]环化反应,最终制备化合物e,具体如下反应式:
。
[0075]
因此,化合物7与化合物c有着本质的区别,即化合物7不能应用于本发明中硅代吲哚啉的合成。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1