一种酰基磷酸酯的合成方法
1.本发明涉及有机合成技术领域,具体涉及一种酰基磷酸酯的合成方法。
背景技术:
2.有机磷酸盐作为结构单元存在于各种常见的天然产物和农药中,具有出色的生物学特性。同时,酰基磷酸酯是极其重要的有机物质之一,已被广泛应用于有机合成中间体,药物和功能材料中。另外,酰基磷酸酯还是许多生化酰化反应的中间体。因此,酰基磷酸酯的合成方法引起了人们的持续关注以及广泛的研究。
3.现有技术的酰基磷酸酯的合成方法主要依赖于酰氯、低温体系、金属催化剂的使用等因素,对环境不友好,合成步骤经济性不高,以及反应底物受限以及可操作性不强的问题。因此,需要开发一种环境友好、成本较低的酰基磷酸酯合成方法。
技术实现要素:
4.本技术的发明人发现,通过以卤化物作为催化剂,能够降低合成酰基磷酸酯的成本、降低反应的环境负担并且以高收率合成酰基磷酸酯。本技术的目的在于提供一种高效、环境友好且低成本的酰基磷酸酯的合成方法。
5.本技术的目的采用以下技术方案实现:
6.本技术提供了一种酰基磷酸酯的合成方法,所述酰基磷酸酯是式(3)所示的化合物,包括:由式(1)所示的过氧化芳基酸酐与式(2)所示的亚磷酸二酯在催化剂的存在下进行酰化反应的步骤,
7.式(1):
8.式(2):
9.式(3):
10.其中,在式(1)~(3)中,r1为取代或未取代的芳基或杂芳基,r2和r3可以相同或不同,并且各自独立地为取代或未取代的具有1~10个碳原子的烷基、取代或未取代的芳基或杂芳基,并且所述催化剂为卤化物。
11.根据本发明的酰基磷酸酯合成方法,能够高效、低成本且环境友好地合成酰基磷酸酯。
12.在一些可以选的实施例中,r1为给电子基团取代的芳基或杂芳基。
13.根据本发明的酰基磷酸酯合成方法,能够以更高的收率合成酰基磷酸酯。
14.在一些可以选的实施例中,r1为未取代的苯基,1~10个碳原子的烷基、卤素、烷氧
基取代的苯基,取代或未取代的萘基和噻吩基中的至少一个,和/或r2和r3各自为未取代的具有1~4个碳原子的烷基和取代或未取代的苯基中的至少一个。
15.根据本发明的酰基磷酸酯合成方法,能够以更高的收率合成酰基磷酸酯。
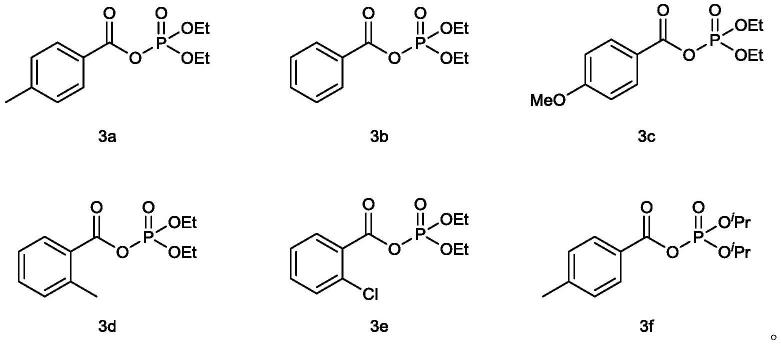
16.在一些可以选的实施例中,所述酰基磷酸酯是式3a至3f中任意一种所示的化合物,
[0017][0018]
在一些可以选的实施例中,所述催化剂为溴化物、碘化物、氯化物中的任意一种或其组合,和/或所述酰化反应的溶剂为乙腈、二氯甲烷、1,2-二氯乙烷和n,n-二甲基甲酰胺中的任意一种或其组合。
[0019]
在一些可以选的实施例中,所述催化剂为溴化钠、溴化锂、溴化钾、溴化铵、四丁基溴化铵、碘化钠和氯化钠中的任意一种或其组合,和/或所述溶剂为乙腈。
[0020]
根据本发明的酰基磷酸酯合成方法,能够进一步降低合成酰基磷酸酯的反应的成本和环境负担。
[0021]
在一些可选的实施例中,式(2)所示的亚磷酸二酯、式(1)所示的过氧化芳基酸酐与所述催化剂的摩尔比为1:(1-3):(0.1-1.5)。
[0022]
根据本发明的酰基磷酸酯合成方法,能够进一步降低合成酰基磷酸酯的反应的成本和环境负担。
[0023]
在一些可选的实施例中,式(2)所示的亚磷酸二酯、式(1)所示的过氧化芳基酸酐与所述催化剂的摩尔比为1:(1.5-2):(0.2-1)。
[0024]
根据本发明的酰基磷酸酯合成方法,能够进一步降低合成酰基磷酸酯的反应的成本。
[0025]
在一些可选的实施例中,所述酰化反应的反应温度为30℃~50℃,和/或所述酰化反应的反应气氛为空气。
[0026]
在一些可选的实施例中,所述酰化反应的反应温度为40℃。
[0027]
根据本发明的酰基磷酸酯合成方法,能够进一步降低合成酰基磷酸酯的成本和环境负担。
具体实施方式
[0028]
下面,结合具体实施方式,对本技术做进一步描述,需要说明的是,在不相冲突的前提下,以下描述的各实施例之间或各技术特征之间可以任意组合形成新的实施例。
[0029]
本发明提供一种酰基磷酸酯的合成方法。具体而言,一种式(3)所示的酰基磷酸酯的合成方法,包括:由式(1)所示的过氧化芳基酸酐与式(2)所示的亚磷酸二酯在催化剂的存在下进行酰化反应的步骤,
[0030]
式(1):
[0031]
式(2):
[0032]
式(3):
[0033]
其中,在式(1)~(3)中,r1为取代或未取代的芳基或杂芳基,r2和r3可以相同或不同,并且各自独立地为取代或未取代的具有1~10个碳原子的烷基、取代或未取代的芳基或杂芳基,并且所述催化剂为卤化物。
[0034]
本发明的酰基磷酸酯合成方法,通过以卤化物代替现有技术的金属催化剂(例如金属na或ag),不仅降低了酰基磷酸酯的合成成本,也降低对环境的负担。
[0035]
对式(1)所示的过氧化芳基酸酐没有特别限制,例如,式(1)中的r1可以是取代或未取代的芳基或杂芳基。优选的,r1可以是取代或未取代的苯基、取代或未取代的萘基、取代或未取代的噻吩基等。对取代的芳基和杂芳基的取代基没有特别限制,但优选为给电子基团,更优选为具有1~10个碳原子的烷基、卤素和烷氧基中至少一种,还更优选为甲基、cl、br和甲氧基中的至少一种,最优选为甲基。
[0036]
对式(2)所示的亚磷酸二酯没有特别限制,例如,式(2)中的r2和r3可以相同或不同,并且r2和r3各自独立地表示具有1~10个碳原子的烷基、取代或未取代的芳基或杂芳基。优选的,r2和r3各自独立地表示具有1~4个碳原子的烷基和取代或未取代的苯基中的至少一个。对取代的芳基和杂芳基的取代基没有特别限制,但优选为具有1~10个碳原子的烷基、卤素和烷氧基中至少一种,更优选为甲基、cl、br和甲氧基中的至少一种,最优选为甲基。
[0037]
优选的,根据本发明的酰基磷酸酯的合成方法,能够合成如下式3a至3f中任意一种所示的化合物。
[0038]
[0039]
对本发明的作为催化剂的卤化物没有特别限制,但优选为溴化物、碘化物、氯化物中的任意一种或其组合,更优选为溴化钠、溴化锂、溴化钾、溴化铵、四丁基溴化铵、碘化钠和氯化钠中的任意一种或其组合,最优选为溴化钠或四丁基溴化铵(tbab)。
[0040]
对催化剂的用量没有特别限制,例如,式(2)所示的亚磷酸二酯、式(1)所示的过氧化芳基酸酐与所述催化剂的摩尔比为1:(1-3):(0.1-1.5),优选的,式(2)所示的亚磷酸二酯、式(1)所示的过氧化芳基酸酐与所述催化剂的摩尔比为1:(1.5-2):(0.2-1)。当催化剂用量过低时,酰化反应无法有效地进行。另一方面,过多催化剂用量并不会带来收率或选择性的进一步提升,反而造成催化剂浪费,增加成本和环境负担。
[0041]
本发明的酰化反应需要在溶剂中进行。溶剂优选为乙腈、二氯甲烷(dcm)、二氯乙烷(dce)和n,n-二甲基甲酰胺(dmf)中的任意一种或其组合,更优选为乙腈。
[0042]
对本发明的酰化反应的反应温度没有特别限定,但优选为30℃~50℃,更优选为35℃~45℃。对本发明的酰化反应的反应气氛没有特别限定,例如可以在空气气氛下进行。
[0043]
实施例
[0044]
以下通过具体实施例来进一步描述本发明,但本发明不限于以下实施例。
[0045]
在实施例中,使用1h-nmr、
13
c-nmr进行化合物的分析和鉴定。
[0046]
《1h-nmr和
13
c-nmr的分析方法》
[0047]
在1h-nmr分析中,使用jeol datum ltd.制造的jnm-ecp400。nmr测量值的积分值为理论值。1h-nmr和
13
c-nmr在室温下在cdcl3上以tms作为内标(400mhz 1
h,101mhz 13
c)在cdcl3中记录,位移(δ)以ppm表示,j值以hz表示。
[0048]
实施例1
[0049]
将过氧化二(4-甲基苯甲酰)(0.80mmol)、亚磷酸二乙酯(0.4mmol)、nabr(20mol%)以及乙腈(4ml)加入到带有搅拌棒的schlenk管(50ml)中,在40℃、空气氛围下封闭搅拌12小时。反应结束后混合物用ch2cl2(3
×
5ml)溶解、过滤并减压浓缩。然后通过tlc技术(10∶1(v/v)石油醚/乙酸乙酯)纯化残余物,得到所需产物(二乙基磷酸)4-甲基苯甲酸酐(3a)。
[0050]
实施例2~6
[0051]
已与实施例1相同的条件,改变式(1)化合物和式(2)化合物种类,分别合成如下所示的混合物3b~3f,产率和表征结果如下。
[0052][0053]
(二乙基磷酸)4-甲基苯甲酸酐(3a):无色油状物。1h-nmr(400mhz,cdcl3)δ7.88(d,j=7.6hz,2h),7.22(d,j=7.5hz,2h),4.30(p,j=7.2hz,4h),2.37(s,3h),1.36(t,j=
7.1hz,6h).
13
c nmr(101mhz,cdcl3)δ160.9(d,j=8.1hz),145.6,130.6,129.4,125.3(d,j=9.1hz),65.2(d,j=5.1hz),21.7,16.0(d,j=7.1hz)。
[0054]
(二乙基磷酸)苯甲酸酐(3b):无色油状物.1h nmr(400mhz,cdcl3)δ8.01(d,j=7.4hz,2h),7.60(t,j=7.4hz,1h),7.44(t,j=7.8hz,2h),4.33(p,j=7.2hz,4h),1.38(t,j=7.1hz,6h).
13
c nmr(101mhz,cdcl3)δ161.0(d,j=8.1hz),134.5,130.6,128.7,128.1(d,j=8.1hz),65.3(d,j=6.1hz),16.0(d,j=7.1hz)。
[0055]
(二乙基磷酸)4-甲氧基苯甲酸酐(3c):无色油状物.1h nmr(400mhz,cdcl3)δ7.59(d,j=7.5hz,1h),7.49(s,1h),7.33(t,j=8.1hz,1h),7.13(dd,j=8.3,2.3hz,1h),4.31(p,j=7.1hz,4h),3.80(s,3h),1.37(t,j=7.2hz,6h).
13
c nmr(101mhz,cdcl3)δ161.7(d,j=9.1hz),160.5,130.5,121.8,115.7,66.1,56.2,16.9。
[0056]
(二乙基磷酸)2-甲基苯甲酸酐(3d):无色油状物.1h nmr(400mhz,cdcl3)δ7.96(d,j=8.4hz,1h),7.46(t,j=7.4hz,1h),7.26(dd,j=8.7,5.5hz,2h),4.34(p,j=7.1hz,4h),2.62(s,3h),1.39(t,j=7.0hz,6h).
13
c nmr(101mhz,cdcl3)δ161.1(d,j=8.1hz),142.8,133.9,132.3,131.9,126.8(d,j=8.1hz),126.2,65.3(d,j=6.1hz),22.2,16.2(d,j=7.1hz)。
[0057]
(二乙基磷酸)2-氯苯甲酸酐(3e):无色油状物.1h nmr(400mhz,cdcl3)δ7.95
–
7.90(m,1h),7.46(dd,j=5.8,2.8hz,2h),7.32(ddd,j=8.3,5.6,3.2hz,1h),4.33(p,j=7.1hz,4h),1.36(t,j=6.6hz,6h).
13
c nmr(101mhz,cdcl3)δ159.5(d,j=8.1hz),135.5,134.5,133.0,131.9,131.3,127.1,65.8(d,j=6.1hz),16.3(d,j=7.1hz)。
[0058]
(二异丙基磷酸)4-甲基苯甲酸酐(3f):无色油状物.1h nmr(400mhz,cdcl3)δ7.89(d,j=8.2hz,2h),7.24
–
7.21(m,2h),4.89(dq,j=12.5,6.2hz,2h),2.38(s,3h),1.37(dd,j=9.0,6.2hz,12h).
13
c nmr(101mhz,cdcl3)δ161.3(d,j=9.1hz),145.7,130.8,129.7,125.9(d,j=8.1hz),74.5(d,j=6.1hz),23.8(dd,j=29.3hz),22.0。
[0059]
实施例7~17和比较例1~3
[0060]
以与实施例1相同的原料,以下表1所示条件合成酰基磷酸酯3a,催化剂的当量数以亚磷酸二乙酯为基准。具体收率如下表1所示。
[0061]
表1
[0062][0063]
[0064]
如表1所示,当使用nai、nacl、nh4br、libr、tbab、kbr作为催化剂时,均表现出较好的酰基磷酸酯收率。然而,当使用i2作为催化剂或不使用催化剂(比较例1~2)时,则无法获得酰基磷酸酯。当使用nabr作为催化剂时,即使以0.2当量的较低量使用时,也能够获得71%的收率(实施例1)。另一方面,当使用dcm、dce和乙腈作为溶剂时,能够获得较高的收率,而当使用dmf时,则只能获得痕量的酰基磷酸酯。反应温度方面,当反应在30℃~50℃反应时,能够获得较高收率,而当反应温度过高或过低时,收率则明显下降。
[0065]
本技术从使用目的上,效能上,进步及新颖性等观点进行阐述,已符合专利法所强调的功能增进及使用要件,本技术以上的说明书,仅为本技术的较佳实施例而已,并非以此局限本技术,因此,凡一切与本技术构造,装置,特征等近似、雷同的,即凡依本技术专利申请范围所作的等同替换或修饰等,皆应属本技术的专利申请保护的范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1