一种2-氨基-4,6-二甲氧基嘧啶的制备方法与流程
1.本发明属于农药中间体的合成技术领域,特别涉及一种2-氨基-4,6-二甲氧基嘧啶的制备方法。
背景技术:
2.氨基-4,6-二甲氧基嘧啶是一种重要的农药中间体,可用于合成磺酰脲类除草剂。磺酰脲类除草剂是一类广谱高效的玉米、水稻、大豆等田地除草剂,具有应用范围广、活性高、药物残留少、用量少、无挥发性及毒性低等特点,因此在世界上被公认为是一种高效、环保的绿色型农药。我国是磺酰脲类除草剂的生产及使用大国,2-氨基-4,6-二甲氧基嘧啶的需求量大约4000~6000吨,其结构式如下:目前,工业上生产2-氨基-4,6-二甲氧基嘧啶的工艺主要有两种。
3.第一种工艺是以硝酸胍和丙二酸二乙酯为起始原料,三氯氧磷为氯化剂,甲醇钠为甲基化试剂,经环合,氯化,甲氧基化得2-氨基-4,6-二甲氧基嘧啶。描述为如下合成路线1:合成路线1但是此工艺中使用了大量三氯氧磷,生产过程中存在安全隐患,且生产过程中会产生大量的有色含磷废水,对水体造成严重污染,废水处理成本高,因此目前该工艺面临减产、停产等问题。
4.第二种工艺,专利文献ep476554、2006mu00152、us4412957、us449510、de2426913a1、cn103159684a和cn105130909a以丙二腈为原料,经过醇解成亚胺盐、氰胺取代、环合三步反应制备2-氨基-4,6-二甲氧基嘧啶。其中,中国专利cn103159684a以丙二腈为原料,在金属氧化物或金属盐酸盐催化下,与甲醇和干燥氯化氢发生加成反应生成1 ,3-二甲氧基丙二亚胺二盐酸盐,然后在缓冲盐(碳酸氢钠和磷酸氢二钠)作用下和单氰胺反应生成3-氨基-3-甲氧基-n-氰基-2-丙脒,最后经酸催化环合或于高沸点体系中加热环合制得2-氨基-4,6-二甲氧基嘧啶。三步反应总收率为61.5~71.1%。以上工艺均可描述为如下合成路线2:
合成路线2尽管合成路线2是目前生产2-氨基-4,6-二甲氧基嘧啶的主要方法,但此工艺路线第一步反应需要在绝对无水条件下制备中间体1,3-二甲氧基丙二亚胺二盐酸盐,收率不高,且常规工艺还需要将此中间体过滤分离,由于反应体系中存在大量氯化氢,中间体对潮湿空气及温度极不稳定,易水解成单酰胺杂质、丙二酰胺、丙二酸等,后期还需要在水性环境中用碳酸氢钠中和、水溶液中氰胺化反应制备3-氨基-3-甲氧基-n-氰基-2-丙脒,后期还需要干燥除水,工艺操作繁琐、且最终收率不高,实际上工业收率仅有65%左右。
技术实现要素:
5.本发明的目的在于提供一种成本低,工艺简单,收率高,废水少的2-氨基-4,6-二甲氧基嘧啶的制备方法,以解决上述背景技术中提出的问题。
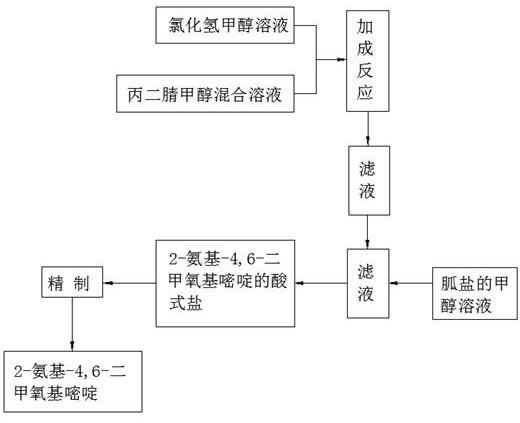
6.为达到上述技术目的,本发明的技术方案:一种2-氨基-4,6-二甲氧基嘧啶的制备方法,包括以下步骤:(1)向无水甲醇中通入干燥的氯化氢气体,制得氯化氢甲醇溶液,通入所述氯化氢气体的温度为-5~10℃,所述氯化氢气体与所述无水甲醇的摩尔比为2.2~2.5: 10~16;(2)将丙二腈和甲醇混合,得到丙二腈甲醇混合溶液,所述甲醇与所述丙二腈的摩尔比为2~3:1;(3)在5~25℃下,将所述丙二腈甲醇混合溶液滴加到所述氯化氢甲醇溶液中,滴加时间为1~3h,滴加完成后,先在5~25℃反应8~12h,再升温至25~45℃继续反应4~8h,反应结束后,过滤,除去氯化铵盐,得滤液;(4)向所述滤液中滴加胍盐的甲醇溶液,所述胍盐与所述丙二腈的摩尔比为1.01~1.1:1,滴加完成后,在5~25℃搅拌反应2~8h,反应结束后,将反应液蒸馏,除去甲醇,得到2-氨基-4,6-二甲氧基嘧啶的酸式盐;(5)向所述2-氨基-4,6-二甲氧基嘧啶的酸式盐中加水进行溶解,然后用无机碱调节ph=7.0~10.0,结晶析出,过滤,水洗,干燥,得到2-氨基-4,6-二甲氧基嘧啶。
7.本发明的合成路线如下:以丙二腈为原料,在氯化氢存在下与甲醇发生加成反应生成1,1,1,3,3,3-六甲氧基丙烷。过滤除去氯化铵盐后,再加入胍盐,反应生成2-氨基-4,6-二甲氧基嘧啶的酸式盐,
再经碱中和得到2-氨基-4,6-二甲氧基嘧啶。
8.通入所述氯化氢气体的温度为-5~10℃。在此温度下,氯化氢气体可以全部溶解于甲醇中,温度过高会导致氯化氢气体挥发,造成污染和损失。
9.步骤(3)中,滴加完成后,先在5~25℃反应8~12h,再升温至25~45℃继续反应4~8h。从反应机理上讲,在5~25℃条件下,首先生成的是脒盐中间体,升温25~40℃后,再继续反应得到六甲氧基丙烷。分两段升温的目的在于让原料丙二腈在低温条件下全部转化为中间体,否则在高温条件下丙二腈与中间体会发生副反应,降低收率。分段反应及中间体结构如下:作为一种改进,步骤(4)中,所述胍盐为盐酸胍、硝酸胍或碳酸胍,优选为盐酸胍。
10.作为一种改进,步骤(4)中,搅拌反应温度为20~25℃,反应时间为4~5h。
11.作为一种改进,步骤(5)中,所述水的加入量为所述丙二腈重量的2~5倍,优选的,所述水的加入量为所述丙二腈重量的4~5倍。
12.作为一种改进,步骤(5)中,所述无机碱为氢氧化钠、碳酸钠、碳酸氢钠、氨水或碳酸铵,优选为氨水。
13.作为一种改进,步骤(5)中,所述ph=7.5~8.2。
14.由于采用上述技术方案,本发明的有益效果:本发明提供的制备方法,为制备2-氨基-4,6-二甲氧基嘧啶提供了一条全新的工艺路线。该制备方法的反应仅为两步反应,解决了传统工艺中多步反应带来的操作复杂性;该制备方法所用溶剂均为甲醇,且无需分离中间产物,操作更加简便、流畅;该制备方法仅在最后一步生产过程中产生废水,而且产生的废水量明显少于传统工艺;该制备方法相比较传统工艺,运行成本大幅降低,降低了生产成本,提高了生产效率,适合工业化生产。
15.本发明以原料丙二腈为起始原料,原料成本低廉,产品总收率最高达到82%以上,产品纯度大于99%,该制备方法的合成路线短,无苛刻条件、操作简单、对环境友好,且溶剂易回收。
附图说明
16.图1是本发明提供的工艺流程图。
具体实施方式
17.下面结合具体实施方式及附图对本发明作进一步的说明。
18.实施例1向1000ml反应器中加入无水甲醇320g(10mol),降温至0℃,通入约81g(2.2mol)氯化氢气体。通气完毕后,缓慢滴加丙二腈(66g,1.0mol)和甲醇(64g,2.0mol)的混合溶液,控制温度在5℃,约2小时滴加完毕。加料完毕后,在5℃继续反应8小时。然后升温至45℃,继续反应4小时。气相色谱检测原料及中间体含量小于0.5%,反应结束。过滤除去产生的氯化铵,
滤液投入下一步反应。
19.将盐酸胍(96.5g,1.01mol)和甲醇(127g)的混合溶液,滴加到上一步反应的滤液中,滴加温度控制在20℃,加料时间为1小时。加料完毕继续反应4小时。液相色谱检测中间体含量小于0.5%,反应结束。
20.后处理:将反应液蒸馏浓缩后,加入264g水溶解。在室温条件下,滴加氢氧化钠,调节ph=7.5,产品结晶析出。过滤、水洗、干燥,得含量为99.5%的2-氨基-4,6-二甲氧基嘧啶产品120.9g,总收率为78%。
21.实施例2向1000ml反应器中加入无水甲醇512g(16mol),降温至-5℃,通入约81g(2.2mol)氯化氢气体。通气完毕后,缓慢滴加丙二腈(66g,1.0mol)和甲醇(64g,2.0mol)的混合溶液,控制温度在15℃,约1小时滴加完毕。加料完毕后,在15℃继续反应10小时。然后升温至25℃,继续反应8小时。气相色谱检测原料及中间体含量小于0.5%,反应结束。过滤除去产生的氯化铵,滤液投入下一步反应。
22.将硝酸胍(123.3g,1.01mol)和甲醇(162g)的混合溶液,滴加到上一步反应的滤液中,滴加温度控制在25℃,加料时间为1小时。加料完毕继续反应2小时。液相色谱检测中间体含量小于0.5%,反应结束。
23.后处理:将反应液蒸馏浓缩后,加330g水溶解。在室温条件下,滴加氨水,调节ph=8.2,产品结晶析出。过滤、水洗、干燥,得含量为99.6%的2-氨基-4,6-二甲氧基嘧啶产品124g,总收率为80%。
24.实施例3向1000ml反应器中加入无水甲醇512g(16mol),降温至5℃,通入约91.3g(2.5mol)氯化氢气体。通气完毕后,缓慢滴加丙二腈(66g,1.0mol)和甲醇(64g,2.0mol)的混合溶液,控制温度在10℃,约2小时滴加完毕。加料完毕后,在10℃继续反应12小时。然后升温至40℃,继续反应6小时。气相色谱检测原料及中间体含量小于0.5%,反应结束。过滤除去产生的氯化铵,滤液投入下一步反应。
25.将盐酸胍(96.5g,1.01mol)和甲醇(127g)的混合溶液,滴加到上一步反应的滤液中,滴加温度控制在15℃,加料时间为1小时。加料完毕继续反应5小时。液相色谱检测中间体含量小于0.5%,反应结束。
26.后处理:将反应液蒸馏浓缩后,加入264g水溶解。在室温条件下,滴加碳酸钠,调节ph=7.0,产品结晶析出。过滤、水洗、干燥,得含量为99.6%的2-氨基-4,6-二甲氧基嘧啶产品127.8g,总收率为82.5%。
27.实施例4向1000ml反应器中加入无水甲醇384g(12mol),降温至10℃,通入约84g(2.3mol)氯化氢气体。通气完毕后,缓慢滴加丙二腈(66g,1.0mol)和甲醇(96g,3.0mol)的混合溶液,控制温度在20℃,约3小时滴加完毕。加料完毕后,在20℃继续反应11小时。然后升温至35℃,继续反应5小时。气相色谱检测原料及中间体含量小于0.5%,反应结束。过滤除去产生的氯化铵,滤液投入下一步反应。
28.将碳酸胍(133.2g,1.1mol)和甲醇(200g)搅拌混合,将上一步反应的滤液滴加到混合溶液中,(由于碳酸胍在甲醇的溶解度较小,所以滴加顺序反过来)滴加温度控制在5
℃,加料时间为1小时。加料完毕继续反应8小时。液相色谱检测中间体含量小于0.5%,反应结束。
29.后处理:将反应液蒸馏浓缩后,加198g水溶解。在室温条件下,滴加氨水,调节ph=9.0,产品结晶析出。过滤、水洗、干燥,得含量为99.5%的2-氨基-4,6-二甲氧基嘧啶产品122.5g,总收率为79%。
30.实施例5向1000ml反应器中加入无水甲醇448g(14mol),降温至7℃,通入约87.6g(2.4mol)氯化氢气体。通气完毕后,缓慢滴加丙二腈(66g,1.0mol)和甲醇(96g,3.0mol)的混合溶液,控制温度在25℃,约2小时滴加完毕。加料完毕后,在25℃继续反应9小时。然后升温至30℃,继续反应7小时。气相色谱检测原料及中间体含量小于0.5%,反应结束。过滤除去产生的氯化铵,滤液投入下一步反应。
31.将盐酸胍(100.3g,1.05mol)和甲醇(132g)的混合溶液,滴加到上一步反应的滤液中,滴加温度控制在10℃,加料时间为1小时。加料完毕继续反应7小时。液相色谱检测中间体含量小于0.5%,反应结束。
32.后处理:将反应液蒸馏浓缩后,加132g水溶解。在室温条件下,滴加氨水,调节ph=10.0,产品结晶析出。过滤、水洗、干燥,得含量为99.6%的2-氨基-4,6-二甲氧基嘧啶产品128.2g,总收率为82.7%。
33.以上所述本发明的具体实施方式,并不构成对本发明保护范围的限定。任何根据本发明的技术构思所做出的各种其他相应的改变与变形,均应包含在本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1