巴豆基氯化钯二聚体的制备方法与流程
1.本发明涉及一种巴豆基氯化钯二聚体的制备方法,属于有机合成技术领域。
背景技术:
2.巴豆基氯化钯二聚体在现代有机合成和实际应用中有着重要的作用,其主要作为催化剂用于suzuki,heck,negishi等偶联反应中,并在液晶材料、光功能配合物等物质的合成中有着极为广泛的应用。
3.现有制备巴豆基氯化钯二聚体的方法是,以氯化钯为起始原料,与氯化钾形成离子状态,再与巴豆基氯络合,生成巴豆基氯化钯二聚体,然后用二氯甲烷萃取,有机相脱溶,干燥得到巴豆基氯化钯二聚体。该方法虽然收率较高,但反应时间太长,通常需要24小时才能完成;而且后处理需要萃取、脱溶等步骤,较为繁琐;还需要耗费大量的水溶解固体原料或作为反应溶剂,因此造成产能低下并伴有废水、废有机溶剂产生。
4.如何缩短巴豆基氯化钯二聚体的生产时间,简化后处理,提高产能,避免产生三废,是巴豆基氯化钯二聚体制备领域亟待解决的技术问题。
技术实现要素:
5.本发明的目的是提供一种新的巴豆基氯化钯二聚体的制备方法,大幅缩短了反应时间,提高了产能,简化了后处理过程,不产生三废,有效解决了现有技术中存在的问题。
6.本发明提供了一种巴豆基氯化钯二聚体的制备方法,其特征在于,用1,5-环辛二烯参与氯化钯和巴豆基氯反应,制备式(i)所示的巴豆基氯化钯二聚体
[0007][0008]
根据本发明的一个具体但非限制性的实施方案,所述的制备方法,包括:在惰性气体保护下,向反应容器中加入氯化钯、1,5-环辛二烯、巴豆基氯和甲苯,在70~100℃的温度下反应0.5小时以上,自然降温到0~30℃,过滤,滤饼干燥,得到式(i)所示的巴豆基氯化钯二聚体。
[0009]
根据本发明的一个具体但非限制性的实施方案,其中,1,5-环辛二烯与氯化钯的摩尔比大于0.5:1。
[0010]
根据本发明的一个具体但非限制性的实施方案,其中,1,5-环辛二烯与氯化钯的摩尔比为1~1.5:1。
[0011]
根据本发明的一个具体但非限制性的实施方案,其中,巴豆基氯与氯化钯的摩尔
比大于1:1。
[0012]
根据本发明的一个具体但非限制性的实施方案,其中,巴豆基氯与氯化钯的摩尔比为1~1.5:1。
[0013]
根据本发明的一个具体但非限制性的实施方案,其中,甲苯为反应溶剂。
[0014]
根据本发明的一个具体但非限制性的实施方案,其中,甲苯与氯化钯的质量比大于3:1。
[0015]
根据本发明的一个具体但非限制性的实施方案,其中,反应中不加甲苯,而使用过量的巴豆基氯充当溶剂。
[0016]
根据本发明的一个具体但非限制性的实施方案,其中,反应时间为2~5小时。
[0017]
本发明的有益效果主要体现在:
[0018]
1.本发明改用1,5-环辛二烯参与氯化钯与巴豆基氯反应,1,5-环辛二烯起催化剂作用,加速了反应进程,大幅缩短了反应时间,使反应时间由现有技术的24小时缩短到3小时左右。
[0019]
2.本发明的辅料全是液体,而产物除了巴豆基氯化钯二聚体是固体以外再无其它固体杂质,因此后处理由现有技术的萃取脱溶等繁琐过程改为了简单的过滤,大大简化了后处理,简便了工艺操作,尤其适合大规模工业化生产。
[0020]
3.本发明的溶剂用量少,同样的溶剂量或同样的反应容器本发明的产能是现有技术的4倍;而且本发明的溶剂无需处理可以直接套用于下次反应,因此没有三废产生,工艺绿色环保。
附图说明
[0021]
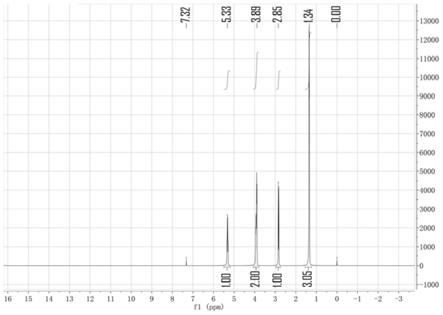
图1是本发明实施例1制备的巴豆基氯化钯二聚体的核磁图谱。
具体实施方式
[0022]
下文提供了具体的实施方式进一步说明本发明,但本发明不仅仅限于以下的实施方式。
[0023]
通常氯化钯与巴豆基氯直接不反应,无产物生成。本技术的发明人经过长期研究发现,如果加入1,5-环辛二烯,在1,5-环辛二烯的参与下,氯化钯与巴豆基氯可以快速反应得到巴豆基氯化钯二聚体。1,5-环辛二烯应是充当了催化剂角色,加速了反应进程,大幅缩短了反应时间,仅3小时就可以完成反应。而且,同样的反应容器,氯化钯的装载量是现有技术的四倍,产能是现有技术的四倍。同时,后处理通过简单的过滤即可;溶剂可以直接套用,无三废,有益效果十分显著。而用1,5-环辛二烯参与氯化钯和巴豆基氯反应制备巴豆基氯化钯二聚体,本技术尚属首次,目前未见文献报道。
[0024]
本发明因此提供一种新的巴豆基氯化钯二聚体的制备方法,用1,5-环辛二烯参与氯化钯和巴豆基氯反应,制备巴豆基氯化钯二聚体。本发明的反应式如下:
[0025][0026]
我们经过研究发现,该反应必须加入1,5-环辛二烯才能进行,仅巴豆基氯与氯化钯不反应。而且我们通过在反应过程中的多个阶段对反应成分进行检测发现,反应过程中始终未有氯化钯与1,5-环辛二烯的络合物存在过,因此我们推测1,5-环辛二烯在该反应中起催化剂作用,加速了反应进程。
[0027]
具体地,本发明制备巴豆基氯化钯二聚体的方法,包括:在惰性气体保护下,向反应容器中加入氯化钯、1,5-环辛二烯、巴豆基氯和甲苯,在70~100℃的温度下反应0.5小时以上,自然降温到0~30℃,过滤,滤饼干燥,得到式(i)所示的巴豆基氯化钯二聚体。
[0028]
其中,1,5-环辛二烯与氯化钯的摩尔比通常大于0.5:1,优选1~1.5:1。
[0029]
巴豆基氯与氯化钯的摩尔比通常大于1:1,优选1~1.5:1。
[0030]
甲苯为反应溶剂,因甲苯与巴豆基氯可以互溶,容易形成均相反应,可以加快反应速度。通常甲苯与氯化钯的质量比大于3:1,优选4~5:1。实际反应中,可以选择性地添加甲苯作为反应溶剂,即可以添加甲苯,也可以不添加甲苯,而使用过量的巴豆基氯充当溶剂。
[0031]
反应时间优选2~5小时。
[0032]
反应后的母液为反应剩余的甲苯、1,5-环辛二烯和巴豆基氯,不需要处理可以直接套用于下次反应,因此没有三废。
[0033]
本发明改用1,5-环辛二烯参与氯化钯与巴豆基氯反应制备巴豆基氯化钯二聚体,大大加快了反应速度,反应时间由现有技术的24小时缩短到2~5小时;本发明的溶剂使用量大幅减少,同样的反应容器中原料氯化钯的装载量大幅提高,是现有技术的四倍,因而产能也是现有技术的四倍;本发明收率可高达98%以上,纯度高达98%。
[0034]
下面结合具体实施例对本发明作进一步阐述,但本发明并不限于以下实施例。
[0035]
上文及下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0036]
上文及下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0037]
实施例1
[0038]
氮气保护下,向10l四口瓶中加入1200g氯化钯,806g 1,5-环辛二烯,675g巴豆基氯,6l甲苯,80℃搅拌3小时,自然降温到20℃,过滤,滤饼干燥得1307g黄色固体巴豆基氯化钯二聚体,滤液直接套用于下次反应,收率98%,纯度98%。图1是实施例1制备的巴豆基氯化钯二聚体的核磁图谱。
[0039]
实施例2
[0040]
氮气保护下,向10l四口瓶中加入1200g氯化钯,806g 1,5-环辛二烯,675g巴豆基氯,6l实施例1的滤液,不足部分用甲苯补充,80℃搅拌2小时,自然降温到20℃,过滤,滤饼干燥得1320g黄色固体巴豆基氯化钯二聚体,滤液直接套用于下次反应,收率99%,纯度98%。
[0041]
实施例3
[0042]
氮气保护下,向10l四口瓶中加入1200g氯化钯,806g 1,5-环辛二烯,6l巴豆基氯,80℃搅拌3小时,自然降温到20℃,过滤,滤饼干燥得1307g黄色固体巴豆基氯化钯二聚体,滤液直接套用于下次反应,收率98%,纯度98%。
[0043]
实施例4
[0044]
氮气保护下,向10l四口瓶中加入1200g氯化钯,366g 1,5-环辛二烯,675g巴豆基氯,6l甲苯,80℃搅拌3小时,自然降温到20℃,过滤,滤饼干燥得1133g黄色固体巴豆基氯化钯二聚体,滤液直接套用于下次反应,收率85%,纯度98%。由于1,5-环辛二烯的用量减少了一半多,1,5-环辛二烯与氯化钯的摩尔比小于0.5:1,故收率比实施例1有所下降。
[0045]
对比例
[0046]
用现有技术方法制备巴豆基氯化钯二聚体
[0047]
氮气保护下,向10l三口瓶中加入300g氯化钯,252g氯化钾,6l水,25℃搅拌1小时,滴加492g巴豆基氯,室温搅拌24小时;然后加5l二氯甲烷萃取,有机相脱溶,干燥得300g黄色固体,收率90%。
[0048]
上述对比例与本发明实施例1比较可知:
[0049]
1.本发明的反应时间由现有技术的24小时缩短到3小时,大幅缩短了反应时间。
[0050]
2.本发明后处理由现有技术的萃取脱溶等繁琐过程改为简单的过滤。原因是本发明辅料全是液体,产物是固体,过滤即可分离;而现有技术工艺产物里有未反应的固体杂质如氯化钾,因此只能通过萃取脱溶的繁琐过程分离。
[0051]
3.本发明的产能是现有技术的4倍。原因是本发明所需的溶剂量少,相同的反应容器,本发明的氯化钯装载量是现有技术的4倍,产能也是现有技术的4倍;而现有技术工艺需要大量水溶解氯化钾或进行反应,所以氯化钯的装载量较少,产能很低。
[0052]
4.现有技术工艺有废水、废有机溶剂产生;而本发明的溶剂可以直接套用于下次反应,所以没有三废,属于绿色环保工艺。
[0053]
以上仅是本发明的具体应用范例,对本发明的保护范围不构成任何限制。凡采用等同变换或者等效替换而形成的技术方案,均落在本发明权利保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1