一类3-吡喃取代特特拉姆酸类化合物及其制备方法和在制备抗炎药物中的应用
1.本发明涉及生物化学技术领域,具体涉及一类3-吡喃取代特特拉姆酸类化合物及其制备方法和在制备抗炎药物中的应用。
背景技术:
2.核因子κb(nuclear factor kappa-b,nf-κb)为一个转录因子蛋白家族,包括rel(c-rel)、 relb、rela/p65、nfκb1 p50/p105和nfκb2 p52/p100。这些蛋白起到二聚化转录因子的作用,可调节基因表达,并可影响到各种不同的生物学过程,包括先天和适应性免疫、炎症、应激反应、b细胞发育和淋巴器官形成。作为早期转录因子,nf-κb的激活不需要新翻译出的蛋白进行调控。因此,可以在第一时间对有害细胞的刺激做出反应。nf-κb通路可被细胞应激、炎症细胞因子、生长因子、紫外射线等激活。nf-κb通路的激活参与了慢性炎症性疾病的发病机制,如哮喘、类风湿性关节炎和炎症性肠病。因此,nf-κb抑制剂具有研究和开发抗炎药物的巨大潜力。
3.特特拉姆酸类化合物是一类具有2,4-吡咯烷二酮母核结构的成分,有时其母核会互变为同分异构体1,5-二氢-4-羟基-2h-吡咯-2-酮。特特拉姆酸类化合物具有多种生物活性,包括抗菌、细胞毒、酶抑制、抗炎等,引起医药学家、合成化学家和合成生物学家的兴趣。但由于特特拉姆酸类化合物结构复杂多样,导致合成步骤繁琐,产率不高,限制了特特拉姆酸类化合物的应用。目前有关于特特拉姆酸类化合物在nf-κb通路的抑制作用的研究较少,尚未发现特特拉姆酸类化合物对在nf-κb通路具有较高活性的抑制作用,因此提供一种高效制备特特拉姆酸类化合物的高效制备方法以及具有高活性nf-κb抑制作用的特特拉姆酸类化合物对慢性炎症性疾病药物开发和治疗有极大的意义。
技术实现要素:
4.本发明克服了上述技术问题,提供可一种深海海洋真菌。
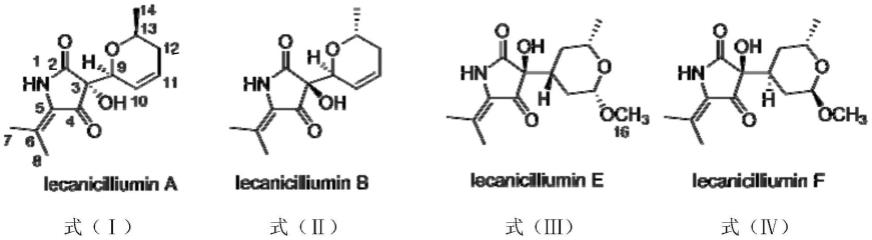
5.本发明请求保护一类3-吡喃取代特特拉姆酸类化合物,所述一类3-吡喃取代特特拉姆酸类化合物为lecanicilliumin a或lecanicilliumin b或lecanicilliumin e或lecanicilliumin f,结构式见式
ⅰ‑
式ⅳ:
[0006][0007]
本发明的另一目的还在于保护一种用于制备上述一类3-吡喃取代特特拉姆酸类
c、化合物 lecanicilliumin d、化合物lecanicilliumin e、化合物lecanicilliuminf;
[0022]
图2为化合物lecanicilliumin a的单晶结构;
[0023]
图3为化合物lecanicilliumin a的氢谱;
[0024]
图4为化合物lecanicilliumin a的碳谱;
[0025]
图5为化合物lecanicilliumin a的hsqc谱图;
[0026]
图6为化合物lecanicilliumin a的1h-1
hcosy谱图;
[0027]
图7为化合物lecanicilliumin a的hmbc谱图;
[0028]
图8为化合物lecanicilliumin a的noesy谱图;
[0029]
图9为化合物lecanicilliumin a的hr-esi-ms谱图;
[0030]
图10为化合物lecanicilliumin b的氢谱;
[0031]
图11为化合物lecanicilliumin b的碳谱;
[0032]
图12为化合物lecanicilliumin b的hsqc谱图;
[0033]
图13为化合物lecanicilliumin b的1h-1
h cosy谱图;
[0034]
图14为化合物lecanicilliumin b的hmbc谱图;
[0035]
图15为化合物lecanicilliumin b的noesy谱图;
[0036]
图16为化合物lecanicilliumin b的hr-esi-ms谱图;
[0037]
图17为化合物lecanicilliumin b的cd测定图谱;具体为化合物lecanicilliumin b在甲醇溶液中的cd测定值与(3s,9s,13r)-2、(3s,9r,13s)-2计算值的比较。σ=0.3ev,uv shift=+7 nm。
[0038]
图18为化合物lecanicilliumin c的氢谱;
[0039]
图19为化合物lecanicilliumin c的碳谱;
[0040]
图20为化合物lecanicilliumin c的hsqc谱图;
[0041]
图21为化合物lecanicilliumin c的1h-1
h cosy谱图;
[0042]
图22为化合物lecanicilliumin c的hmbc谱图;
[0043]
图23为化合物lecanicilliumin c的noesy谱图;
[0044]
图24为化合物lecanicilliumin c的hr-esi-ms谱图;
[0045]
图25为化合物lecanicilliumin d的氢谱;
[0046]
图26为化合物lecanicilliumin d的碳谱;
[0047]
图27为化合物lecanicilliumin d的hsqc谱图;
[0048]
图28为化合物lecanicilliumin d的1h-1
h cosy谱图;
[0049]
图29为化合物lecanicilliumin d的hmbc谱图;
[0050]
图30为化合物lecanicilliumin d的noesy谱图;
[0051]
图31为化合物lecanicilliumin d的hr-esi-ms谱图;
[0052]
图32为化合物lecanicilliumin d的cd测定图谱;具体为化合物lecanicilliumin d在甲醇溶液中的cd测定值与(3s,9s,13s)-4、(3s,9r,13r)-4、(3r,9r,13r)-4、(3r,9s,13s)
ꢀ‑
4计算值的比较。σ=0.3ev,uv shift=-1nm;
[0053]
图33为化合物lecanicilliumin e的氢谱;
[0054]
图34为化合物lecanicilliumin e的碳谱;
[0055]
图35为化合物lecanicilliumin e的hsqc谱图;
[0056]
图36为化合物lecanicilliumin e的cosy谱图;
[0057]
图37为化合物lecanicilliumin e的hmbc谱图;
[0058]
图38为化合物lecanicilliumin e的noesy谱图;
[0059]
图39为化合物lecanicilliumin e的一维noesy差谱(1.98ppm);
[0060]
图40为化合物lecanicilliumin e的一维noesy差谱(4.24ppm);
[0061]
图41为化合物lecanicilliumin e的hr-esi-ms谱图;
[0062]
图42为化合物lecanicilliumin f的氢谱;
[0063]
图43为化合物lecanicilliumin f的碳谱;
[0064]
图44为化合物lecanicilliumin f的hsqc谱图;
[0065]
图45为化合物lecanicilliumin f的1h-1
h cosy谱图;
[0066]
图46为化合物lecanicilliumin f的hmbc谱图;
[0067]
图47为化合物lecanicilliumin f的noesy谱图;
[0068]
图48为化合物lecanicilliumin f的hr-esi-ms谱图;
具体实施方式
[0069]
下面结合实施例和试验对本发明作进一步说明。
[0070]
实施例1
[0071]
深海海洋真菌(lecanicillium fusisporum)gximd00542,其是从一株马里亚纳海沟沉积物(采样点141
°
57’e,10
°
51’n,水深5467米)中采样得到,菌株2021年11月30日保存于广东省微生物菌种保藏中心,地址:广州市先烈中路100号大院59号楼5楼,其保藏编号为gdmcc no:62091。
[0072]
实施例2
[0073]
一类3-吡喃取代特特拉姆酸类化合物的制备:
[0074]
1、发酵培养基的配置:将葡萄糖10克,甘露醇20克,麦芽糖20克,谷氨酸钠10克,磷酸二氢钾0.5克,硫酸镁0.5克,酵母提取物3克,海盐30克,水定容至1升。按此比例配置发酵用培养基。将该培养基分装于约100个1升的三角瓶中,每瓶约300毫升培养基。在高压灭菌锅中121℃灭菌20分钟。
[0075]
2、发酵:将深海海洋真菌(lecanicillium fusisporum)gximd00542在合适的平板培养基上活化。用无菌竹签挑取活化后的菌丝体至约5瓶上述准备的三角瓶中,在恒温摇床上在 25℃,180rpm条件下培养48小时,进行种子液的培养。上述100瓶培养基按每瓶5毫升加入种子液。在25℃条件下静置培养30天,得到发酵产物。
[0076]
3、提取:过滤发酵产物,分为发酵液和菌丝体。发酵液用大孔树脂吸附。收取大孔树脂,用20%的甲醇冲洗去除培养基成分,再用甲醇(也可以用乙醇)洗脱,甲醇洗脱液减压浓缩后得到菌液提取物(发酵液也可以用乙酸乙酯、二氯甲烷或氯仿萃取)。菌丝体粉碎后用丙酮(也可以用乙酸乙酯)浸泡提取,减压浓缩后得到菌体提取物。合并菌液提取物和菌体提取物得到总的发酵提取物。
[0077]
4、分离:将提取物用正相硅胶(100-200目)干法拌样,按1:20的重量比例装入正相硅胶层析柱(200-300目),常温常压条件下,依次用100:0,90:10,80:20,70:30, 60:40比例的二氯甲烷-丙酮洗脱层析柱。洗脱液减压浓缩后,通过薄层层析色谱检测合并类似组分,
共得到15个组分(f1_f15)。f9组分(0.41克)经过反相硅胶柱色谱分离。反相硅胶140克,洗脱系统为乙腈-水10%-40%,得到20个亚组分(sf9-1_sf9-20)。sf9-14经过高效液相制备(检测波长254nm,流速为3毫升/分钟,色谱柱为ymc 250
×
10mm,5μm。流动相为40:60的甲醇-水),出峰时间28.6分钟得到化合物lecanicilliumin c(40毫克),出峰时间33.6分钟得到化合物lecanicilliumin d(14毫克);sf9-11经过高效液相制备(检测波长254nm,流速为3毫升/分钟,色谱柱为ymc 250
×
10mm,5μm。流动相为18:82 的乙腈-水),出峰时间30.3分钟得到化合物lecanicilliumin e(8毫克);sf9-13经过高效液相制备(检测波长254nm,流速为3毫升/分钟,色谱柱为ymc 250
×
10mm,5μm。流动相为40:60的甲醇-水),出峰时间28.9分钟得到化合物lecanicilliumin f(11毫克);sf9-15 经过高效液相制备(检测波长254nm,流速为3毫升/分钟,色谱柱为ymc 250
×
10mm,5μm。流动相为22:78的乙腈-水),出峰时间35.8分钟得到化合物lecanicilliumin b(8毫克),出峰时间38.2分钟得到化合物lecanicilliumin a(5毫克)。
[0078]
化合物的结构解析如下:
[0079]
化合物lecanicilliumin a为浅褐色固体,高分辨质谱给出准分子离子峰m/z 274.1063[m+na]
+
(计算值:274.1055),结合核磁氢谱、碳谱数据(参见下表1和表2),确定化合物lecanicilliumin a的分子式为c
13h17
no4。参见图1-图9,hsqc谱图表明该化合物含有3个甲基ch3,1个亚甲基ch2,4个次甲基ch,5个季碳c。hmbc谱图中nh-1(10.36,1h,brs)与c-2(171.45,c),c-2(74.31,c),c-3(197.12,c),c-6(128.96,c)间的远程相关,ch
3-7(1.77,3h,s),ch
3-8(2.02,3h,s)与c-5(120.24,c),c-6(128.96,c)间的远程相关表明存在5-异丙亚基-2,4-吡咯烷二酮结构(1.huang zh,nong xh,liang x,qish.tetrahedron,2018,74,2620-2620),即特特拉姆酸类结构。h-hcosy谱图中h-10(5.82, 1h,dd,j=10.2,2.1hz)与h-9(4.30,1h,s)、h-11(5.97,1h,d,j=10.2hz)相关,h-12(1.65,1h,m;1.90,1h,m)与h-11、h-13(3.52,1h,ddd,j=7.8,7.6,4.6 hz)相关,h-13与ch
3-14(0.99,3h,d,j=6.1hz)相关(参见图1);根据化合物lecanicilliumina的不饱和度、分子式及氢谱、碳谱数据,表明除了特特拉姆酸类结构外,该化合物还存在一个环状结构,即2-甲基-3,6-二氢吡喃基团。hmbc谱图h-9(4.30,1h,s)与c-3(197.12, c)、c4相关,oh-3(6.32,1h,br s)与c-9相关,表明2-甲基-3,6-二氢吡喃基团连接在 5-异丙亚基-2,4-吡咯烷二酮结构的3位(参见图1)。化合物的立体构型通过x-单晶衍射光谱确定为(3r,9s,13s)。化合物lecanicilliumin a在甲醇溶剂中得到斜方晶,晶体大小为 0.16
×
0.08
×
0.08立方毫米,射线由铜靶cukα产生,flack常数为0.03(参见图2)。晶体数据保存于英国剑桥晶体数据中心(ccdc),编号为2092073。
[0080]
化合物lecanicilliumin b也是浅褐色固体,参见图1以及图10-图17,高分辨质谱给出的准分子离子峰m/z 274.1059[m+na]
+
(计算值:274.1055)、氢谱、碳谱数据(参见表1和表2)表明化合物lecanicilliumin b与化合物lecanicilliumin a具有相同的平面结构。两个化合物在核磁数据上的最大区别是c-2化学位移(化合物ecanicilliumin a:171.45;化合物 lecanicilliumin b:169.70)和c-4化学位移(化合物lecanicilliumin a:197.12;化合物lecanicilliumin b:199.36)。noesy谱图h-9(4.23,1h,m)与ch
3-14(0.99,3h,d,j=6.1 hz)相关,表明它们在2-甲基-3,6-二氢吡喃基团的同侧。化合物lecanicilliumin b的绝对构型通过与比较圆二色谱(cd)数据和计算圆二色谱(ecd)技术确定。根据化合物
lecanicilliumina的cd谱图与文献报道(huang zh,nong xh,liang x,qi sh.tetrahedron,2018,74, 2620-2620),当c-3为r构型时,cd图谱在200-210处呈正的cotton效应,230-240nm处呈负的cotton效应,而化合物lecanicilliumin b的cd图谱在在200-210处呈负的cotton效应,230-240nm处呈正的cotton效应,表明化合物lecanicilliumin b的c-3位构型为s。因此化合物lecanicilliumin b的绝对构型为3s,9s,13r或者3s,9r,13s。使用gaussian 16软件对 (3s,9s,13r)-2或者(3s,9r,13s)-2进行ecd计算。计算结果表明(参见图17),化合物lecanicilliumin b的cd图谱与(3s,9s,13r)-2计算cd谱图相似度为92%,与(3s,9r,13s)
ꢀ‑
2计算cd谱图相似度为49%,从而确定化合物lecanicilliumin b的绝对构型为3s,9r,13s。
[0081]
化合物lecanicilliumin c为白色粉末,参见图1以及图18-图24,高分辨质谱数据m/z 252.1241[m+h]
+
(计算值252.1236)表明化合物lecanicilliumin c具有和化合物lecanicilliumina、化合物lecanicilliumin b相同的分子式c
13h17
no4,核磁氢谱(参见表1)、碳谱数据(参见表2)与化合物lecanicilliumin a、化合物lecanicilliumin b类似,但2-甲基-3,6-二氢吡喃基团上双键的化学位移和耦合常数发生较大变化。hmbc谱图(参见图1)h-10(4.51,d,j=6.4 hz)与c-9(37.73,ch)、c-11(145.33,ch)、c-14(29.70,ch2)相关,h-11(6.35,dd,j =6.4,2.3hz)与c-9(37.73,ch)、c-10(97.81,ch)、c-13(71.20,ch)相关,表明2
‑ꢀ
甲基-二氢吡喃基团的双键位置由c4-c5重排到了c5-c6,即2-甲基-3,4-二氢吡喃基团。hmbc 谱图h-9(2.64,1h,ddd,j=11.4,5.8,2.0hz)与c-2(171.35,c)、c-3(75.14,c)、 c-4(199.04,c)相关,表明5-异丙亚基-2,4-吡咯烷二酮结构连接在2-甲基-3,4-二氢吡喃基团的c4位,从而确定了化合物lecanicilliumin c的平面结构。化合物lecanicilliumin c的 cd图谱在200-210处呈负的cotton效应,在240nm处呈正的cotton效应,表明化合物 lecanicilliumin c的c-3位为s构型。noesy谱图h-9与h-13相关,表明它们在2-甲基-3,4
‑ꢀ
二氢吡喃基团的同侧。因此化合物lecanicilliumin c的绝对构型为3s,9s,13r或3s,9r,13s 者。化合物lecanicilliumin c的绝对构型通过gaussian16软件计算碳谱确定。对(3s,9s,13r)
ꢀ‑
3和(3s,9r,13s)-3分别采用b97-2/pcsseg-1计算,计算结果通过dp4+拟合,均显示其绝对构型计算结果为(3s,9s,13r)。
[0082]
化合物lecanicilliumin d为白色固体,参见图1以及图25-图32,高分辨质谱数据及相似的氢谱、碳谱数据表明化合物lecanicilliumin d具有和化合物lecanicilliumin c相同的平面结构。核磁数据差异最大在c-9位、c-13位和c-14位的化学位移(表1和表2)。化合物 lecanicilliumin d的立体结构通过noesy谱图、cd数据和ecd计算确定。noesy谱图h-9与ch
3-15 相关,表明它们在2-甲基-3,4-二氢吡喃基团的同侧。因此化合物lecanicilliumin d的绝对构型为3s,9s,13s、3s,9r,13r、3r,9r,13r或者3r,9s,13s。ecd计算结果表明(图32),化合物lecanicilliumin d的实测cd谱图与(3s,9s,13s)-4、(3s,9r,13r)-4计算cd谱图均相近。通过gaussian16软件对(3s,9s,13s)-4和(3s,9r,13r)-4分别采用b97-2/pcsseg-1 计算碳谱,计算结果通过dp4+拟合,均显示其绝对构型计算结果为(3s,9s,13s),综合ecd 计算结果和碳谱计算结果,从而确定其绝对构型为(3s,9s,13s)-4。
[0083]
参见图1以及图33-图41,化合物lecanicilliumin e的高分辨质谱数据m/z 306.1319[m+ na]
+
和核磁数据(参见表1和表2),表明分子式为c
14h21
no5(计算值:306.1317)。核磁数据表明,化合物lecanicilliumin e同样存在5-异丙亚基-2,4-吡咯烷二
酮结构(参见表1和表 2)。h-hcosy谱图数据表明h-9与h-10、h-14相关,h-11与h-10相关,h-13与h-14,ch
3-15 相关,hmbc谱图数据h-11与c-13相关,och
3-16与c-11相关,表明存在一个2-甲氧基-6 甲基四氢吡喃基团。hmbc谱图数据h-9与c-2、c-3和c-4相关,表明2-甲氧基-6甲基四氢吡喃基团连接在5-异丙亚基-2,4-吡咯烷二酮母核的c-3位。化合物lecanicilliumin e的相对构型通过noesy和选择性一维noe谱图确定。noesy谱图h-9与h-13相关;选择性一维noe 谱图h-9与h-11、h-13相关,h-11与h-9、h-13相关,表明它们在2-甲氧基-6甲基四氢吡喃基团的同侧。化合物lecanicilliumin e的cd图谱在200-210处呈负的cotton效应,在 230-240nm处呈正的cotton效应确定c-3位为s构型。因此化合物lecanicilliumin e的绝对构型可能为(3s,9r,11s,13s)或者(3s,9s,11r,13r)。对两种构型分别采用b97-2/pcsseg-1 计算碳谱,计算结果通过dp4+拟合,均显示其绝对构型计算结果为(3s,9r,11s,13s),综合 ecd计算结果和碳谱计算结果,从而确定其绝对构型为(3s,9r,11s,13s)-5。
[0084]
参见图1以及图42-图48,化合物lecanicilliumin f与化合物lecanicilliumin e具有相同的高分辨质谱数据m/z 306.1319[m+na]
+
和类似的核磁数据(参见表1和表2),它们具有相同的分子式和类似的化学结构。核磁数据差异最大在c-9位、c-11位和c-13位的化学位移(参见表1和表2)。hmbc和h-hcosy谱图相关数据表明,化合物lecanicilliumin f与化合物lecanicilliumin e具有相同的平面结构。noesy谱图h-9与h-11相关,h-13与och
3-16 相关,表明h-9,h-13,ch
3-15在2-甲氧基-6甲基四氢吡喃基团平面的同侧,h-13和och
3-16 在平面的另一侧。化合物lecanicilliumin f的cd图谱在200-210处呈负的cotton效应,在 240nm处呈正的cotton效应,表明化合物lecanicilliumin f的c-3位为s构型。因此,化合物 lecanicilliumin f的绝对构型可能是(3s,9s,11r,13s)或者(3s,9r,11s,13r)。对两种构型分别采用b97-2/pcsseg-1计算碳谱,计算结果通过dp4+拟合,均显示其绝对构型计算结果为(3s,9s,11r,13s),综合ecd计算结果和碳谱计算结果,从而确定其绝对构型为 (3s,9s,11r,13s)-6。
[0085]
表1.化合物lecanicilliumin a-f的氢谱数据(600mhz,dmso-d6)
[0086][0087]
表2.化合物lecanicilliumin a-f的氢谱数据(150mhz,dmso-d6)
[0088]
[0089][0090]
根据以上的结构分析可知,化合物lecanicilliumin a-f的结构式如下,其中 lecanicilliumin a的结构式见式(ⅰ),化合物lecanicilliumin b的结构式见式(ⅱ)、化合物lecanicilliumin c的结构式见式(
ⅴ
)、化合物lecanicilliumin d的结构式见式 (ⅵ)、化合物化合物lecanicilliumin e的结构式见式(ⅲ)、化合物lecanicilliumin f 的结构式见式(ⅳ):
[0091][0092]
实施例3化合物的抑制活性测定:
[0093]
测定方法:采用nf-κb荧光素酶报告基因法在细胞水平测定了化合物lecanicilliumin a-f 抑制脂多糖(lps)诱导的nf-κb升高活性。细胞系为稳定转染nf-κb荧光素酶报告基因的小鼠单核巨噬细胞raw264.7。细胞用不同浓度梯度的化合物和阳性对照药物共同培养4小时,加入脂多糖(浓度为100ng/ml),再温育6小时,用荧光素酶检测系统测定。
[0094]
实验结果:化合物lecanicilliumin a,化合物lecanicilliumin b,化合物lecanicilliumin e,和化合物lecanicilliumin f抑制nf-κb半数抑制浓度分别为18.49
±
1.21μmol/l,25.81
±
1.30 μmol/l,23.10
±
1.26μmol/l,24.70
±
1.19μmol/l和26.52
±
1.12μmol/l,而化合物lecanicilliuminc,化合物lecanicilliumin d无抑制活性。活性测定结果表明,部分新化合物能够显著降低脂多糖诱导的nf-κb升高,可以用于制备抗炎药物。
[0095]
上述说明是针对本发明较佳可行实施例的详细说明,但实施例并非用以限定本发明的专利申请范围,凡本发明所提示的技术精神下所完成的同等变化或修饰变更,均应属于本发明所涵盖专利范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1