一种阿尔兹海默症PET-tau示踪剂的制备方法与流程
一种阿尔兹海默症pet-tau示踪剂的制备方法
技术领域
1.本发明涉及化合物制备技术领域,尤其涉及一种阿尔兹海默症pet-tau示踪剂的制备方法。
背景技术:
2.阿尔茨海默症(alzheimer disease,ad)是一种最常见的、不可逆的、与年龄相关的神经退行性大脑疾病,该疾病会导致病人缓慢丧失记忆和诱发其他精神疾病。随着全球老龄化日益显著,ad的发病人数在逐年增加。早发现、早诊断、早干预是改善患者预后、提高生活质量的重要措施,因此ad的病理学特异性诊断工具的开发一直受到全世界科学家的关注。
3.聚集的tau蛋白是神经退行性疾病如ad的病理生理学的主要神经病理学底物。7-(6-氟吡啶-3-基)-5h-吡啶基[4,3-b]吲哚(记为化合物t807)是一种吲哚类药物,对tau蛋白具有较高的亲和力和选择性,同时具有合适的体内药物代谢动力学特征,被广泛应用于ad的高度选择性和特异性pet-tau示踪剂。
[0004]
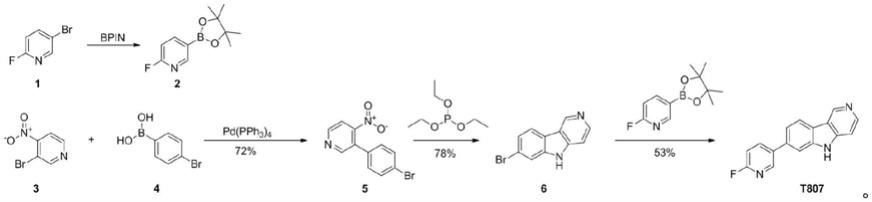
有报道(bioorganic&medicinal chemistry letters 25(2015)2953-2957)采用如下路线制备化合物t807:
[0005][0006]
但是采用上述方法制备化合物t807时,是以化合物3为原料,该原料不易获得,需要采用双氧水、硝酸以及硫酸等危险品经多个步骤制备得到,而且在制备化合物t807时,三个步骤需要用到钯催化剂,导致化合物t807生产成本高。此外,采用上述方法制备化合物t807,收率较低,总收率仅为29.76%。
技术实现要素:
[0007]
本发明的目的在于提供一种阿尔兹海默症pet-tau示踪剂的制备方法,本发明提供的方法原料易获得,只在一个步骤中使用钯催化剂,目标化合物生产成本低,且收率高。
[0008]
为了实现上述发明目的,本发明提供以下技术方案:
[0009]
本明提供了一种阿尔兹海默症pet-tau示踪剂的制备方法,所述阿尔兹海默症pet-tau示踪剂为7-(6-氟吡啶-3-基)-5h-吡啶基[4,3-b]吲哚,包括以下步骤:
[0010]
将化合物i、化合物ii、催化剂与第一有机溶剂混合,在保护气氛中进行缩合反应,得到化合物iii;所述催化剂为对甲苯磺酸或冰乙酸;
[0011]
将所述化合物iii与多聚磷酸混合,在保护气氛中进行缩合成环反应,得到化合物
iv;
[0012]
将所述化合物iv、联硼酸频那醇酯、乙酸钾、三(二亚苄-base丙酮)二钯、2-二环己基膦-2',4',6'-三异丙基联苯与第二有机溶剂混合,在保护气氛中进行第一偶联反应,得到中间产物体系;将所述中间产物体系、5-氯-2-氟吡啶的第二有机溶剂溶液与k3po4的水溶液混合,在保护气氛中进行第二偶联反应,得到阿尔兹海默症pet-tau示踪剂;
[0013]
所述化合物i、化合物ii、化合物iii和化合物iv的结构式依次如式i、式ii、式iii和式iv所示:
[0014][0015]
其中,所述r1为-cl、-br或-i;
[0016]
所述r2和r3独立地选自c1~c20的脂肪烃基。
[0017]
优选地,所述化合物i与化合物ii的摩尔比为1:(1~2),所述催化剂与化合物i的用量比为(0.01~1.00)g:(20~200)mmol。
[0018]
优选地,所述缩合反应的温度为100~130℃,时间为3~6h。
[0019]
优选地,所述多聚磷酸与化合物i的用量比为1mmol:(1.0~1.5)g。
[0020]
优选地,所述缩合成环反应的温度为120~160℃,时间为1~3h。
[0021]
优选地,所述化合物iv与联硼酸频那醇酯的摩尔比为1:(1.8~2.2),所述乙酸钾与化合物iv的摩尔比为(1.8~2.2):1,所述(二亚苄-base丙酮)二钯、2-二环己基膦-2',4',6'-三异丙基联苯与化合物iv的摩尔比为(0.18~0.22):(0.38~0.42):10。
[0022]
优选地,所述第一偶联反应的温度为90~130℃,时间为2~5h。
[0023]
优选地,所述5-氯-2-氟吡啶与化合物iv的摩尔比为(0.8~1.2):1,所述k3po4与化合物iv的摩尔比为(4~6):1。
[0024]
优选地,所述第二偶联反应的温度为90~130℃,时间为4~8h。
[0025]
优选地,所述脂肪烃基为烷基。
[0026]
本发明提供了一种阿尔兹海默症pet-tau示踪剂的制备方法,本发明以化合物i为起始原料,原料易得且价格低,有利于降低目标化合物生产成本;本发明提供的方法反应路线短,核心路线只有三个步骤,只在第三个步骤中使用到钯催化剂,且该步骤使用一锅法合成,有利于进一步降低成本。同时,本发明提供的方法产物收率较现有技术明显提高,实施例的结果显示,采用本发明提供的方法,产物收率最高可达73.2%。
附图说明
[0027]
图1为实施例1制备的7-溴-5h-吡啶[4,3-b]吲哚的1h nmr图;
[0028]
图2为实施例1制备的化合物t807的1h nmr图。
具体实施方式
[0029]
本发明提供了一种阿尔兹海默症pet-tau示踪剂的制备方法,所述阿尔兹海默症
pet-tau示踪剂为7-(6-氟吡啶-3-基)-5h-吡啶基[4,3-b]吲哚,包括以下步骤:
[0030]
将化合物i、化合物ii、催化剂与第一有机溶剂混合,在保护气氛中进行缩合反应,得到化合物iii;所述催化剂为对甲苯磺酸或冰乙酸;
[0031]
将所述化合物iii与多聚磷酸混合,在保护气氛中进行缩合成环反应,得到化合物iv;
[0032]
将所述化合物iv、联硼酸频那醇酯、乙酸钾、三(二亚苄-base丙酮)二钯、2-二环己基膦-2',4',6'-三异丙基联苯与第二有机溶剂混合,在保护气氛中进行第一偶联反应,得到中间产物体系;将所述中间产物体系、5-氯-2-氟吡啶的第二有机溶剂溶液与k3po4的水溶液混合,在保护气氛中进行第二偶联反应,得到阿尔兹海默症pet-tau示踪剂;
[0033]
所述化合物i、化合物ii、化合物iii和化合物iv的结构式依次如式i、式ii、式iii和式iv所示:
[0034][0035]
其中,所述r1为-cl、-br或-i;
[0036]
所述r2和r3独立地选自c1~c20的脂肪烃基。
[0037]
本发明将化合物i、化合物ii、催化剂与第一有机溶剂混合,在保护气氛中进行缩合反应,得到化合物iii;所述催化剂为对甲苯磺酸或冰乙酸。在本发明中,所述化合物i的结构式如式i所示,所述式i中r1为-cl、-br或-i,优选为-br,此时所述化合物i为6-溴吲哚-3-甲醛。在本发明中,所述化合物ii的结构式如式ii所示,所述式i中r2和r3独立地选自c1~c20的脂肪烃基,所述c1~c20的脂肪烃基优选为c1~c20的烷基,更优选为c1~c10的烷基,进一步优选为c1~c5的烷基,更进一步优选为甲基或乙基,此时所述化合物ii为氨基乙醛缩二甲醇或氨基乙醛缩二乙醇。
[0038]
在本发明中,所述化合物i与化合物ii的摩尔比优选为1:(1~2),更优选为1:(1.2~1.7)。在本发明中,所述催化剂与化合物i的用量比优选为(0.01~1.00)g:(20~200)mmol;具体的,当所述催化剂为对甲苯磺酸时,所述对甲苯磺酸与化合物i的用量比优选为(0.1~1.0)g:22.3mmol,更优选为0.1g:22.3mmol;当所述催化剂为冰乙酸时,所述冰乙酸与化合物i的用量比优选为(0.01~0.10)g:200mmol,更优选为0.1g:200mmol。在本发明中,所述第一有机溶剂优选包括苯或甲苯,更优选为甲苯;所述第一有机溶剂的用量保证缩合反应顺利进行即可,本发明对此不作特殊限定;所述第一有机溶剂使用前优选进行干燥。本发明对所述化合物i、化合物ii、催化剂与第一有机溶剂的混合方式没有特殊限定,能够将各组分充分混合即可。
[0039]
本发明对提供所述保护气氛的保护气体种类没有特殊限定,具体可以为氮气。在本发明中,所述缩合反应的温度优选为100~130℃,更优选为110~120℃,具体可以为体系回流温度;所述缩合反应的时间优选为3~6h,更优选为3~4h;所述缩合反应优选在搅拌条件下进行。在本发明的实施例中,具体是在带机械搅拌及分水器的三口烧瓶中进行所述缩
合反应,至所述分水器中有机相澄清,停止反应。
[0040]
所述缩合反应后,本发明优选将所得产物体系趁热减压浓缩,得到棕黑色油状物,为化合物iii的粗品,无需提纯直接进行下一步反应。
[0041]
得到化合物iii后,本发明将所述化合物iii与多聚磷酸混合,在保护气氛中进行缩合成环反应,得到化合物iv。在本发明中,所述多聚磷酸(ppa)优选为无水多聚磷酸;所述多聚磷酸的作用是脱水。在本发明中,所述多聚磷酸与化合物i的用量比优选为1mmol:(1.0~1.5)g,更优选为1mmol:(1.2~1.4)g。在本发明中,所述化合物iii与多聚磷酸混合优选是向所述多聚磷酸中分批加入化合物iii,具体是将所述多聚磷酸加热至80~100℃后停止加热,分批次加入所述化合物iii(具体为化合物iii的粗品),所述化合物iii加料的整个过程,体系温度控制在80~110℃,加料完毕后开始加热进行所述缩合成环反应。
[0042]
在本发明中,所述缩合成环反应的温度优选为120~160℃,更优选为150~160℃;所述缩合成环反应的时间优选为1~3h,更优选为2~3h;所述缩合成环反应优选在搅拌条件下进行。在本发明的实施例中,具体是在带机械搅拌的四口瓶中进行所述缩合成环反应。本发明优选通过tlc监测反应进程。
[0043]
所述缩合成环反应后,本发明优选将所得产物体系降温至50~80℃,在搅拌条件下滴加第一氢氧化钠水溶液,至体系的ph值为8~10,过滤,将所得滤饼用水洗涤后得到化合物iv粗品;将所述化合物iv粗品与盐酸混合进行成盐反应,之后用二氯甲烷进行萃取,将萃取后所得水相与第二氢氧化钠水溶液混合进行中和反应,之后过滤,将所得滤饼用甲醇洗涤后干燥,得到浅棕黄色固体,即为7-溴-5h-吡啶[4,3-b]吲哚。在本发明中,所述第一氢氧化钠水溶液的浓度优选为20~50wt%,更优选为40~50wt%。在本发明中,所述盐酸的浓度优选为1~2mol/l,所述盐酸的用量以保证化合物iv充分成盐为基准;所述成盐反应的温度优选为80~110℃,更优选为90~100℃;所述成盐反应的时间优选为2~6h,更优选为3~4h。在本发明中,所述第二氢氧化钠水溶液的浓度优选为20~50wt%,更优选为40~50wt%;本发明优选向萃取后的水相中加入碎冰,在0~5℃、搅拌条件下滴加第二氢氧化钠水溶液,至体系ph值为9~10,进行中和反应;所述中和反应的时间优选为2~5h,更优选为2~3h。本发明优选通过利用盐酸使化合物iv成盐,从而能够溶于水中,便于利用二氯甲烷反萃,提取出杂质和副产物;之后再利用氢氧化钠水溶液将水相中化合物iv的盐酸盐中和,从水中析出游离态的化合物iv,达到提纯效果。
[0044]
得到化合物iv后,本发明将所述化合物iv、联硼酸频那醇酯、乙酸钾、三(二亚苄-base丙酮)二钯、2-二环己基膦-2',4',6'-三异丙基联苯与第二有机溶剂混合,在保护气氛中进行第一偶联反应,得到中间产物体系。在本发明中,所述化合物iv与联硼酸频那醇酯(bpin)的摩尔比优选为1:(1.8~2.2),更优选为1:2。在本发明中,所述乙酸钾的作用是催化剂,所述乙酸钾与化合物iv的摩尔比优选为(1.8~2.2):1,更优选为2:1。在本发明中,所述三(二亚苄-base丙酮)二钯(pd2dba3)为催化剂,2-二环己基膦-2',4',6'-三异丙基联苯(x-phos)为配体催化剂,用于提高pd2dba3的催化活性;所述pd2dba3、x-phos与化合物iv的摩尔比优选为(0.18~0.22):(0.38~0.42):10,更优选为0.1:0.4:10。在本发明中,所述第二有机溶剂优选包括二氧六环、苯或甲苯,更优选为二氧六环;所述第二有机溶剂的用量以保证第一偶联反应顺利进行即可,本发明对此不作特殊限定。本发明对所述化合物iv、联硼酸频那醇酯、乙酸钾、三(二亚苄-base丙酮)二钯、2-二环己基膦-2',4',6'-三异丙基联苯
与第二有机溶剂混合的方式没有特殊限定,保证各组分充分混合即可。
[0045]
在本发明中,所述第一偶联反应的温度优选为90~130℃,更优选为100~110℃,具体可以为体系回流温度;所述第一偶联反应的时间优选为2~5h,更优选为3~4h。在本发明中,所述第一偶联反应时,化合物iv与bpin偶联生成硼酸频那醇酯中间体。本发明优选通过tlc监测反应进程。
[0046]
在本发明中,所述第一偶联反应后,所得中间产物体系无需进行任何后处理,直接与5-氯-2-氟吡啶的第二有机溶剂溶液和k3po4的水溶液混合,在保护气氛中进行第二偶联反应,得到阿尔兹海默症pet-tau示踪剂。在本发明中,所述5-氯-2-氟吡啶与化合物iv的摩尔比优选为(0.8~1.2):1,更优选为1:1;所述5-氯-2-氟吡啶的第二有机溶剂溶液的浓度优选为0.8~1.2mmol/ml,更优选为1.0mmol/ml。在本发明中,所述k3po4为催化剂,所述k3po4与化合物iv的摩尔比优选为(4~6):1,更优选为5:1;所述k3po4的水溶液的浓度优选为4~6mol/l,更优选为5mol/l。本发明对所述中间产物体系、5-氯-2-氟吡啶的第二有机溶剂溶液与k3po4的水溶液混合的方式没有特殊限定,保证各组分充分混合即可。
[0047]
在本发明中,所述第二偶联反应(suzuki反应)的温度优选为90~130℃,更优选为100~110℃,具体可以为体系回流温度;所述第二偶联反应的时间优选为4~8h,更优选为5~6h。本发明优选通过tlc监测反应进程。
[0048]
所述第二偶联反应后,本发明优选将所得产物体系冷却后减压蒸馏去除溶剂,剩余物经水洗涤后采用二氯甲烷进行萃取,所得有机相经无水硫酸镁干燥后过滤,所得滤液经减压蒸馏去除溶剂,剩余物用甲醇-二氯甲烷混合溶剂进行重结晶,得到的白色固体为阿尔兹海默症pet-tau示踪剂(化合物t807)。在本发明中,所述甲醇-二氯甲烷混合溶剂中甲醇与二氯甲烷的体积比优选为2:8~4:6,更优选为2:8。
[0049]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0050]
实施例1
[0051]
(1)制备n-((6-溴-1h-吲哚-3-基)亚甲基)-2,2-二甲氧基乙胺,反应式如下:
[0052][0053]
于带机械搅拌及分水器的三口烧瓶中加入干燥的甲苯40ml、6-溴吲哚-3-甲醛5.0g(22.3mmol)、氨基乙醛缩二甲醇4.0g(38.1mmol)和对甲苯磺酸0.1g,在n2保护下,于110℃进行回流分水3h,此时分水器中有机相澄清,停止反应;将所得产物体系趁热减压浓缩,得到棕黑色油状物,为n-((6-溴-1h-吲哚-3-基)亚甲基)-2,2-二甲氧基乙胺粗品,无需提纯直接进行下一步反应。
[0054]
(2)制备7-溴-5h-吡啶[4,3-b]吲哚,反应式如下:
[0055][0056]
于带机械搅拌的四口瓶中加入无水多聚磷酸(ppa)30g,在n2保护下加热至80℃后停止加热,分批次加入所述棕黑色油状物,所述棕黑色油状物加料的整个过程,体系温度控制在100~110℃(反应剧烈放热,故加料过程中不需要额外加热),加料完毕后开始加热,使体系在150~160℃条件下进行缩合成环反应3h,tlc监测反应完全;
[0057]
反应结束后,将所得产物体系降温至80℃,在搅拌速率为72rpm的条件下逐滴加入浓度为50wt%的氢氧化钠水溶液,至体系ph值为9,过滤,所得滤饼用去离子水洗涤3次,得到7-溴-5h-吡啶[4,3-b]吲哚粗品;
[0058]
将所述7-溴-5h-吡啶[4,3-b]吲哚粗品加入到200ml浓度为1mol/l的盐酸中,在90℃条件下搅拌4h使7-溴-5h-吡啶[4,3-b]吲哚成盐,之后用二氯甲烷萃取3次(100ml
×
3),回收萃取后的有机相,所述有机相中残留固体为少量未反应的6-溴吲哚-3-甲醛,可以回收使用;向萃取后的水相中加入碎冰,在0~5℃、搅拌条件下逐滴加入浓度为50wt%的氢氧化钠水溶液,至体系ph值为9,继续搅拌2h,之后过滤,所得滤饼用甲醇洗涤3次,干燥后所得浅棕黄色固体即为7-溴-5h-吡啶[4,3-b]吲哚,产量为4.3g,纯度为98.6%;步骤(1)和步骤(2)的总收率为78.0%。
[0059]
图1为产物的1hnmr图,表征数据如下:
[0060]1h nmr(dmso-d6):d 7.41(dd,j=2.0,8.5hz,1h,ar-h),7.50(dd,j=1.0,5.5hz,1h,ar-h),7.77(d,j=2.0hz,1h,ar-h),8.1(d,j=8.5hz,1h,ar-h),8.4(d,j=5.5hz,1h,ar-h),9.36(s,1h,ar-h),11.82(s,1h,nh)。
[0061]
(3)制备7-(6-氟吡啶-3-基)-5h-吡啶基[4,3-b]吲哚(化合物t807),反应式如下:
[0062][0063]
在三口烧瓶中依次加入7-溴-5h-吡啶[4,3-b]吲哚(2.47g,10mmol)、联硼酸频那醇酯(bpin,5.1g,20mmol)、乙酸钾(1.96g,20mmol)、三(二亚苄-base丙酮)二钯(0)(pd2dba3,91.5mg,0.1mmol)、2-二环己基膦-2',4',6'-三异丙基联苯(x-phos,0.19g,0.4mmol)和40ml二氧六环,在n2保护下,于110℃回流反应3h,tlc监测反应完全;
[0064]
反应结束后,通过注射器将5-氯-2-氟吡啶(1.31g,10mmol)的二氧六环(10ml)溶液和10ml浓度为5mol/l的k3po4水溶液加入到所得反应体系中,于110℃回流反应6h,tlc监测反应完全;
[0065]
反应结束后,将所得产物体系冷却后减压除去二氧六环,剩余物用蒸馏水洗涤3次,之后用二氯甲烷萃取3次(30ml
×
3),萃取所得有机相用无水硫酸镁干燥,滤掉干燥剂,所得滤液经减压蒸馏回收二氯甲烷,剩余物用甲醇-二氯甲烷混合溶剂(甲醇与二氯甲烷的体积比为2:8)进行重结晶,得到的白色固体为化合物t807,产量为2.47g,纯度为99.3%,收
率为93.8%;步骤(1)、步骤(2)和步骤(3)的总收率为73.2%。
[0066]
图2为产物的1h nmr图;表征数据如下:
[0067]
rf=0.34(1:12meoh/ch2cl2),mp》300℃;
[0068]1h nmr(dmso-d6):d 7.33(dd,j=2.8,8.4hz,1h,ar-h),7.50(d,j=5.2hz,1h,ar-h),7.61(dd,j=1.2,8.4hz,1h,ar-h),7.85(d,1h,ar-h),8.33
–
8.46(m,3h,ar-h),8.66(d,j=5.6hz,1h,ar-h),9.39(s,1h,ar-h),11.88(brs,1h,nh)。
[0069]
实施例2
[0070]
(1)制备n-((6-溴-1h-吲哚-3-基)亚甲基)-2,2-二乙氧基乙胺,反应式如下:
[0071][0072]
于带机械搅拌及分水器的三口烧瓶中加入干燥的甲苯600ml、6-溴吲哚-3-甲醛44.8g(0.2mol)、氨基乙醛缩二乙醇32.0g(0.24mol)和冰乙酸0.1g,在n2保护下,于110℃进行回流分水4h,此时分水器中有机相澄清,停止反应;将所得产物体系趁热减压浓缩,得到棕黑色油状物,为n-((6-溴-1h-吲哚-3-基)亚甲基)-2,2-二乙氧基乙胺粗品,无需提纯直接进行下一步反应。
[0073]
(2)制备7-溴-5h-吡啶[4,3-b]吲哚,反应式如下:
[0074][0075]
于带机械搅拌的四口瓶中加入无水多聚磷酸(ppa)250g,在n2保护下加热至80℃后停止加热,分批次加入所述棕黑色油状物,所述棕黑色油状物加料的整个过程,体系温度控制在100~110℃(反应剧烈放热,故加料过程中不需要额外加热),加料完毕后开始加热,使体系在150~160℃条件下进行缩合成环反应3h,tlc监测反应完全;
[0076]
反应结束后,将所得产物体系降温至80℃,在快速搅拌(搅拌速率为70rpm)条件下逐滴加入浓度为50wt%的氢氧化钠水溶液,至体系ph值为9,过滤,所得滤饼用去离子水洗涤3次,得到7-溴-5h-吡啶[4,3-b]吲哚粗品;
[0077]
将所述7-溴-5h-吡啶[4,3-b]吲哚粗品加入到1l浓度为2mol/l的盐酸中,在90℃条件下搅拌3h使7-溴-5h-吡啶[4,3-b]吲哚成盐,之后用二氯甲烷萃取3次(300ml
×
3),回收萃取后的有机相,所述有机相中残留固体为少量未反应的6-溴吲哚-3-甲醛,可以回收使用;向萃取后的水相中加入碎冰,在0~5℃、搅拌条件下逐滴加入浓度为50wt%的氢氧化钠水溶液,至体系ph值为9,继续搅拌3h,过滤,所得滤饼用甲醇洗涤3次,干燥后所得浅棕黄色
固体即为7-溴-5h-吡啶[4,3-b]吲哚,产量为36.4g,纯度为98.3%;步骤(1)和步骤(2)的总收率为73.7%。
[0078]
(3)制备7-(6-氟吡啶-3-基)-5h-吡啶基[4,3-b]吲哚(化合物t807),反应式如下:
[0079][0080]
在三口烧瓶中依次加入7-溴-5h-吡啶[4,3-b]吲哚(24.7g,0.1mol)、联硼酸频那醇酯(bpin,51g,0.2mol)、乙酸钾(19.6g,0.2mol)、三(二亚苄-base丙酮)二钯(0)(pd2dba3,0.92g,1.0mmol)、2-二环己基膦-2',4',6'-三异丙基联苯(x-phos,1.9g,4mmol)和500ml二氧六环,在n2保护下,于110℃回流反应4h,tlc监测反应完全;
[0081]
反应结束后,通过注射器将5-氯-2-氟吡啶(13.1g,0.1mol)的二氧六环(100ml)溶液和80ml浓度为5mol/l的k3po4水溶液加入到所得反应体系中,于110℃回流反应5h(,tlc监测反应完全;
[0082]
反应结束后,将所得产物体系冷却后减压除去二氧六环,剩余物用蒸馏水洗涤3次,之后用二氯甲烷萃取3次(200ml
×
3),萃取所得有机相用无水硫酸镁干燥,滤掉干燥剂,所得滤液经减压蒸馏回收二氯甲烷,剩余物用甲醇-二氯甲烷混合溶剂(甲醇与二氯甲烷的体积比为2:8)进行重结晶,得到的白色固体为化合物t807,产量为25.48g,纯度为99.1%,收率为96.8%;步骤(1)、步骤(2)和步骤(3)的总收率为71.3%。
[0083]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1