新型烷基氨类化合物或盐、异构体、其制备方法及用途与流程
1.本发明涉及药物化学领域,具体涉及新型烷基氨类化合物或其盐、异构体、其制备方法及其药物组合物在制备治疗和预防中的用途,尤其在疼痛、药物成瘾、阿片药物增效、精神病等的治疗中的用途。
背景技术:
2.目前世界疼痛的发病率大约为35-45%,老年人的发病率更高,约为75%-90%,疼痛已是全球排位第三的治疗领域,市场巨大。阿片类药物是目前最强且常用的中枢镇痛药,过去的研究已经表明阿片类药物(如可待因、吗啡、羟考酮、芬太尼)可有效止痛。它们与脑、脊髓以及胃肠道中的阿片受体结合从而减轻疼痛感。正是由于阿片类药物止痛效果显著,其滥用已经成为美国一个主要的公共健康问题;每天有78人因阿片类药物过量而死亡。因此临床上急需新靶点的强效镇痛药。
3.sigma受体是独特的非阿片类受体,广泛分布于中枢神经系统,σ1受体与所有主要中枢神经系统疾病的病理生理学有关,包括情绪障碍(焦虑和抑郁)、精神病和精神分裂症,以及药物成瘾和疼痛。sigma 1受体拮抗剂s1ra(制备例1化合物)可增强阿片类镇痛剂的镇痛活性,增效作用可被甲基纳曲酮、σ1受体激动剂、纳曲酮所拮抗。s1ra还能单独用于治疗疼痛,如在急性神经痛、坐骨神经痛、糖尿病性神经痛、炎症性疼痛及内脏疼痛等动物模型中表现出较强镇痛作用。其次,s1ra没有阿片样镇痛药的成瘾性,耐受性好。目前s1ra 在欧洲完成了5个临床ⅱ期研究,其中用于治疗奥沙利铂诱发的周围性神经病变的验证性临床表明,s1ra疗效不显著;术后疼痛,术前先给予400mg s1ra,术后再给予吗啡(联用), s1ra组吗啡消耗量增加了,但恶心,呕吐和头晕的不良反应降低,止吐药用量减少,平均疼痛强度减弱,并具有统计学意义;术后神经痛,28天降低了疼痛程度,达到统计学意义;糖尿病性神经性疼痛,带状疱疹后神经痛实验结果无统计学意义。因此s1ra在多个临床ⅱ期效果并不佳,仅在长期用药的术后神经痛镇痛效果明显,但其耐受性好。总体临床结果表明,s1ra不管是单用或是与吗啡联用的镇痛作用还有待提高。
4.通过对s1ra的结构分析,本发明人设计了全新的镇痛药物,并进行多种模型的体内药效试验验证后,意外发现了镇痛作用比s1ra强数量级倍数的镇痛药物,在低剂量下,在小鼠醋酸扭体试验、大鼠热板试验及大鼠热辐射甩尾试验中,独立使用都表现出非常强的镇痛作用。
技术实现要素:
5.本发明化合物为一种新型的镇痛药,实施例化合物在动物体内表现出良好的镇痛作用。在小鼠醋酸扭体实验中,腹腔注射给予1mg/kg本发明化合物时,仍然有极强的镇痛作用;并且实施例化合物5和实施例化合物10在大鼠热板镇痛试验(1mg/kg,i.v.)和大鼠热辐射甩尾试验(4mg/kg,i.v.)中亦表现出强大的镇痛作用,然而同等剂量的s1ra在同等试验条件下却与空白对照组相当,未表现镇痛作用。
6.一方面,本发明涉及式ⅰ的化合物或式ⅰ所示化合物的立体异构体、几何异构体、互变异构体、氮氧化合物、水合物、溶剂化合物、代谢产物、药学上可接受的盐或前药,
[0007][0008]
其中,环a为取代或未取代的c5-10的碳环基、取代或未取代的c5-10芳基、5-10个原子组成的取代或未取代的杂环基或5-10个原子组成的取代或未取代的杂芳基;
[0009]
x为n或c;
[0010]
y为o或s或nh;
[0011]
z为n或c,虚线代表与其相连的c形成双键或单键;
[0012]
r1选自氢、氘、取代或未取代的烷基、取代或未取代的环烷基、取代或未取代的烯基、取代或未取代的芳基、取代或未取代的芳烷基、取代或未取代的杂环基、取代或未取代的杂环基烷基、-cor4、-c(o)or4、-c(o)nr4r5、-c-nr4、-oc(o)r4、-nr4c(o) r5、-n=cr4 r5、-nr4r5、-no2、-cn、-sr4、-or4或卤素;
[0013]
r2和r3独立地选自氢、氘、取代或未取代的烷基、取代或未取代的环烷基、取代或未取代的烯基、取代或未取代的芳基、取代或未取代的芳烷基、取代或未取代的杂环基、取代或未取代的杂环基烷基,或者与它们相连的氮原子合起来形成取代或未取代的杂环基基团;
[0014]
n选自0、1、2、3、4、5或6;
[0015]
r4和r5各自独立地选自氢、氘、取代或未取代的烷基、取代或未取代的环烷基、取代或未取代的烯基、取代或未取代的芳基、取代或未取代的杂环基、取代或未取代的芳烷基、取代或未取代的杂环烷基,或者合起来它们形成稠环体系。
[0016]
进一步地,如式ⅰ所示的化合物或式ⅰ所示化合物的立体异构体、几何异构体、互变异构体、氮氧化合物、水合物、溶剂化合物、代谢产物、药学上可接受的盐或前药,
[0017][0018]
其中,环a为c
5-6
的取代或未取代的碳环基、c
5-6
取代或未取代的芳基、5-6个原子组成的取代或未取代的杂环基;
[0019]
y为o或s;
[0020]
z为n或c,虚线代表与其相连的c形成双键或单键;
[0021]
r1选自氢、取代或未取代的c
1-12
烷基、取代或未取代的c
4-12
环烷基、取代或未取代的 c
2-12
烯基、取代或未取代的c
5-12
芳基、取代或未取代的c
5-12
杂环基、取代或未取代的烷氧基、取代或未取代的烷硫基、取代或未取代的氨基或卤素;
[0022]
r2和r3独立地选自氢、氘、取代或未取代的c
1-12
烷基、取代或未取代的c
4-12
环烷基、取代或未取代的c
2-12
烯基、取代或未取代的c
5-10
芳基、取代或未取代的c
5-10
杂环基,或者与它们相连的氮原子合起来形成取代或未取代的3元至15元杂环基基团;
[0023]
n选自0、1、2、3、4、5、6。
[0024]
进一步地,如式ⅰ所述的化合物、或其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药,具有通式ⅰ'所示的结构,
[0025][0026]
其中,b、c、d选自c或n,且b、c、d至多两个为n;
[0027]
r1选自氢、取代或未取代的c
1-12
烷基、取代或未取代的c
4-12
环烷基、取代或未取代的 c
2-12
烯基、取代或未取代的c
5-12
芳基、取代或未取代的c
5-12
杂环基、取代或未取代的烷氧基、取代或未取代的烷硫基、取代或未取代的氨基或卤素;
[0028]
r2和r3独立地选自氢、氘、取代或未取代的c
1-12
烷基、取代或未取代的c
4-12
环烷基、取代或未取代的c
2-12
烯基、取代或未取代的c
5-10
芳基、取代或未取代的c
5-10
杂环基,或者与它们相连的氮原子合起来形成取代或未取代的3元至15元杂环基基团;
[0029]
n选自0、1、2或3;
[0030]
其中,所述取代基选自一个或多个的氘、卤素、羟基、c
1-c6烷基、c
1-c6烷氧基、c
1-c6烷硫基、胺基、c
1-c6烷胺基、c
1-c6烷酰胺基、c
5-c
10
芳基、c
5-c
10
芳烷基、c
5-c
10
杂环基、 no2、cn、cf3,或者合起来它们形成稠环体系。
[0031]
进一步地,如式ⅰ所述的化合物、或其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药,具有通式ⅰ"所示结构:
[0032][0033]
y为o或s;
[0034]
z为n或c,虚线代表与其相连的c形成双键或单键;
[0035]
r1为取代或未取代的环丙基、取代或未取代的环已基、取代或未取代的环戊基、取代或未取代的苯基、取代或未取代的吡啶基、取代或未取代的嘧啶基、取代或未取代的噻吩基、取代或未取代的噻唑基、取代或未取代的吡咯基、取代或未取代的呋喃环、取代或未取代的萘基、取代或未取代的喹啉基、取代或未取代的二苯并噻吩基、取代或未取代的烷氧基、取代或未取代的丙基、取代或未取代的烯丙基;
[0036]
r2和r3独立地选自氢、氘、甲基、乙基、丙基、丁基、取代或未取代的芳甲基、取代或未取代的芳乙基、取代或未取代的芳杂甲基、取代或未取代的芳杂乙基,或r2和r3与它们相连的氮原子合起来形成取代或未取代的吗啉环基、取代或未取代的哌嗪环基、取代或未取代的吡咯烷基、取代或未取代的哌啶基、取代或未取代的六氢嘧啶基、取代或未取代的咪唑基、取代或未取代的硫代吗啉基、取代或未取代的氮杂环辛烷基、取代或未取代的氮杂环庚烷基;
[0037]
其中,所述取代基选自一个或多个的氘、卤素、羟基、甲基、乙基、环丙基、叔丁基、甲氧基、乙氧基、环丙氧基、叔丁氧基、甲硫基、乙硫基、环丙硫基、叔丁硫基、胺基、甲胺基、乙胺基、环丙胺基、叔丁胺基、甲酰胺基、乙酰胺基、环丙酰胺基、叔丁酰胺基、no2、 cn、cf3。
[0038]
n选自1或2。
[0039]
进一步地,如式ⅰ、ⅰ'或ⅰ"所述的化合物、其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药:
[0040]
r1选自取代或未取代的苯基、取代或未取代的萘基、取代或未取代的吡啶基、取代或未取代的联苯基、取代或未取代的二苯并噻吩基、取代或未取代的喹啉基;其中,所述取代基选自一个或多个的氘、卤素、羟基、甲基、乙基、环丙基、叔丁基、甲氧基、乙氧基、环丙氧基、叔丁氧基、甲硫基、乙硫基、环丙硫基、叔丁硫基、胺基、甲胺基、乙胺基、环丙胺基、叔丁胺基、甲酰胺基、乙酰胺基、环丙酰胺基、叔丁酰胺基、no2、cn、cf3;
[0041]
r2和r3独立地选自甲基、乙基或r2和r3与它们相连的氮原子合起来形成吗啉环基、哌嗪环基、哌啶基、氮杂环庚烷基;
[0042]
n选自1或2。
[0043]
进一步地,如式ⅰ、ⅰ'或ⅰ"所述的化合物、其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药:
[0044]
r1选自2-萘基、2-氯苯基、4-氟苯基、2-甲基-4-氟苯基、苯基、3,5-二氟苯基、3,4
二氯苯基、3,4二氟苯基、4-(三氟甲氧基)苯基、环己基、烯丙基、烯丁基、正丙基、异丙基、正丁基、乙基、环丙基、4-甲氧基苯基、2-甲氧基苯基、2-(三氟甲氧基)苯基、2-甲硫基苯基、 4-二苯并噻吩基、4-氰苯基、4-喹啉基、3-羟基丙基、3-氟丙基、联苯基、3-羟基苯基、2-氨基苯基、吡啶基、甲氧基、羟基、硝基、氨基、2-氯-4-氟苯基、4-氟-2-甲氧基、4-氟-2-(三氟甲基)苯基、4-氯-2-甲氧基苯基、2,4-二氯苯基、2,4-二氟苯基、2,4-双(三氟甲基)苯基、4
‑ꢀ
氯-2-(三氟甲基)苯基;
[0045]
r2和r3独立地选自甲基、乙基;
[0046]
n选自1或2;
[0047]
其中r1为硝基、氨基、甲氧基、羟基时,取代在7位。
[0048]
进一步地,如式ⅰ、ⅰ'或ⅰ"所述的化合物、其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药:
[0049]
r1选自2-萘基、2-氯苯基、4-氟苯基、2-甲基-4-氟苯基、苯基、3,5-二氟苯基、3,4二氯苯基、3,4二氟苯基、4-(三氟甲氧基)苯基、环己基、烯丙基、烯丁基、正丙基、异丙基、正丁基、乙基、环丙基、4-甲氧基苯基、2-甲氧基苯基、2-(三氟甲氧基)苯基、2-甲硫基苯基、 4-二苯并噻吩基、4-氰苯基、4-喹啉基、3-羟基丙基、3-氟丙基、联苯基、3-羟基苯基、2-氨基苯基、溴、吡啶基、甲基、甲氧基、羟基、硝基、氨基、2-氯-4-氟苯基、4-氟-2-甲氧基、 4-氟-2-(三氟甲基)苯基、4-氯-2-甲氧基苯基、2,4-二氯苯基、2,4-二氟苯基、2,4-双(三氟甲基)苯基、4-氯-2-(三氟甲基)苯基;
[0050]
r2和r3与它们相连的氮原子合起来形成吗啉环基;
[0051]
n选自1或2;
[0052]
其中r1为硝基、氨基、甲氧基、溴、羟基、甲基、吡啶时,取代在7位。
[0053]
进一步地,如式ⅰ、ⅰ'或ⅰ"所述的化合物、其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药:
[0054]
r1选自2-萘基、2-氯苯基、4-氟苯基、2-甲基-4-氟苯基、苯基、3,5-二氟苯基、3,4二氯苯基、3,4二氟苯基、4-(三氟甲氧基)苯基、环己基、烯丙基、烯丁基、正丙基、异丙基、正丁基、乙基、环丙基、4-甲氧基苯基、2-甲氧基苯基、2-(三氟甲氧基)苯基、2-甲硫基苯基、 4-二苯并噻吩基、4-氰苯基、4-喹啉基、3-羟基丙基、3-氟丙基、联苯基、3-羟基苯基、2-氨基苯基、溴、吡啶基、甲基、甲氧基、羟基、硝基、氨基、2-氯-4-氟苯基、4-氟-2-甲氧基、 4-氟-2-(三氟甲基)苯基、4-氯-2-甲氧基苯基、2,4-二氯苯基、2,4-二氟苯基、2,4-双(三氟甲基)苯基、4-氯-2-(三氟甲基)苯基;
[0055]
r2和r3与它们相连的氮原子合起来形成哌嗪环基;
[0056]
n选自1或2;
[0057]
其中,其中r1为溴时,取代在5位。
[0058]
进一步地,如式ⅰ、ⅰ'或ⅰ"所述的化合物、其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药:
[0059]
r1选自2-萘基、2-氯苯基、4-氟苯基、2-甲基-4-氟苯基、苯基、3,5-二氟苯基、3,4二氯苯基、3,4二氟苯基、4-(三氟甲氧基)苯基、环己基、烯丙基、烯丁基、正丙基、异丙基、正丁基、乙基、环丙基、4-甲氧基苯基、2-甲氧基苯基、2-(三氟甲氧基)苯基、2-甲硫基苯基、 4-二苯并噻吩基、4-氰苯基、4-喹啉基、3-羟基丙基、3-氟丙基、联苯基、3-羟基苯基、2-氨基
苯基、溴、吡啶基、甲基、3-吡啶基、甲氧基、硝基、氨基、2-氯-4-氟苯基、4-氟-2-甲氧基、4-氟-2-(三氟甲基)苯基、4-氯-2-甲氧基苯基、2,4-二氯苯基、2,4-二氟苯基、2,4-双(三氟甲基)苯基、4-氯-2-(三氟甲基)苯基;
[0060]
r2和r3与它们相连的氮原子合起来形成哌啶基;
[0061]
n选自1或2;
[0062]
其中r1为硝基、氨基、甲氧基、溴、甲基时,取代在7位。
[0063]
进一步地,如式ⅰ、ⅰ'或ⅰ"所述的化合物、其立体异构体或互变异构体、或其药学上可接受的盐、水合物、溶剂化物或前药:
[0064]
r1选自2-萘基、2-氯苯基、4-氟苯基、2-甲基-4-氟苯基、苯基、3,5-二氟苯基、3,4二氯苯基、3,4二氟苯基、4-(三氟甲氧基)苯基、环己基、烯丙基、烯丁基、正丙基、异丙基、正丁基、乙基、环丙基、4-甲氧基苯基、2-甲氧基苯基、2-(三氟甲氧基)苯基、2-甲硫基苯基、 4-二苯并噻吩基、4-氰苯基、4-喹啉基、3-羟基丙基、3-氟丙基、联苯基、3-羟基苯基、2-氨基苯基、溴、吡啶基、甲基、甲氧基、羟基、硝基、氨基、2-氯-4-氟苯基、4-氟-2-甲氧基、 4-氟-2-(三氟甲基)苯基、4-氯-2-甲氧基苯基、2,4-二氯苯基、2,4-二氟苯基、2,4-双(三氟甲基)苯基、4-氯-2-(三氟甲基)苯基;
[0065]
r2和r3与它们相连的氮原子合起来形成氮杂环庚烷基;
[0066]
n选自1或2。
[0067]
进一步的,本发明涉及式ⅰ、ⅰ'、ⅰ"的化合物,优选化合物包括,但不限于选自下列化合物或其药学上可接受的盐或其前药:
[0068]
[0069]
[0070]
[0071][0072]
进一步的,本发明涉及式ⅰ、ⅰ'、ⅰ"的化合物,优选化合物包括,但不限于选自下列化合物或其药学上可接受的盐或其前药:
[0073]
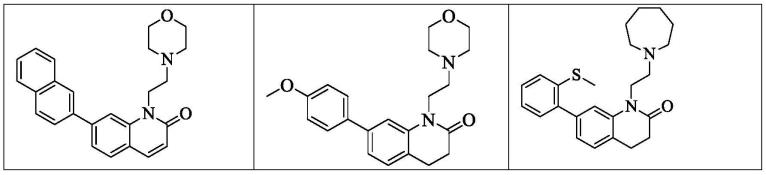
1-(2-吗啉代乙基)-7-(萘-2-基)喹啉-2(1h)-酮
[0074]
1-(2-吗啉代乙基)-7-(萘-2-基)喹喔啉-2(1h)-酮
[0075]
7-(2-氯苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0076]
7-溴-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0077]
1-(2-吗啉代乙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)-酮
[0078]
7-(4-氟苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0079]
7-(4-氟-2-甲基苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0080]
1-(2-吗啉代乙基)-7-苯基-3,4-二氢喹啉-2(1h)-酮
[0081]
7-(3,5-二氟苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0082]
7-(3,4-二氯苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0083]
7-(3,4-二氟苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0084]
1-(2-吗啉代乙基)-7-(4-(三氟甲氧基)苯基)-3,4-二氢喹啉-2(1h)-酮
[0085]
7-(4-甲氧基苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0086]
7-(2-甲氧基苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0087]
1-(2-吗啉代乙基)-7-(2-(三氟甲氧基)苯基)-3,4-二氢喹啉-2(1h)-酮
[0088]
7-(2-(甲硫基)苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0089]
1-(2-吗啉代乙基)-7-(吡啶-3-基)-3,4-二氢喹啉-2(1h)-酮
[0090]
7-(3,4-二氯苯基)-1-[2-(哌啶-1-基)乙基]-3,4-二氢喹啉-2(1h)-酮
[0091]
7-(萘-2-基)-1-(2-(哌啶-1-基)乙基)-3,4-二氢喹啉-2(1h)-酮
[0092]
7-[2-(甲硫基)苯基]-1-[2-(哌啶-1-基)乙基]-3,4-二氢喹啉-2(1h)-酮
[0093]
7-(二苯并[b,d]噻吩-3-基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0094]
4-[1-(2-吗啉代乙基)-2-氧代-1,2,3,4-四氢喹啉-7-基]苄腈
[0095]
1'-(2-吗啉代乙基)-3',4'-二氢-[-4,7'-双喹啉]-2'(1'h)-酮
[0096]
1-(3-吗啉代乙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)-酮
[0097]
1-[2-(氮杂环庚烷-1-基)乙基]-7-(2-甲硫苯基)-3,4-二氢喹啉-2(1h)-酮
[0098]
1-[2-(氮杂环庚烷-1-基)乙基]-6-甲氧基-3,4-二氢喹啉-2(1h)-酮
[0099]
7-(2-氯苯基)-1-[2-(二甲基氨基)乙基]-3,4-二氢喹啉-2(1h)-酮
[0100]
6-(3,4-二氯苯基)-1-(2-(哌啶-1-基)乙基)-3,4-二氢喹啉-2(1h)-酮
[0101]
7-(3-羟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0102]
7-(3-氟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0103]
6-(3-羟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0104]
6-(3-氟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0105]
7-([1,1'-联苯]-3-基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0106]
7-([1,1'-联苯]-4-基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮
[0107]
1-(2-吗啉代乙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)-硫酮。
[0108]
进一步的,本发明涉及式ⅰ、ⅰ'、ⅰ"的化合物或其药学上可接受的盐、异构体,其特征在于,所述化合物中的氢可被一个或多个氘所取代。
[0109]
进一步的,本发明涉及式ⅰ的化合物、关键中间体及其盐的制备方法。
[0110]
方法一:
[0111][0112]
化合物ii与有机硼酸(ⅲ)或有机硼酸酯(iv)在1,4-dioxane、dmf、dmso、dme、 1,4-dioxane/h2o等溶剂中,pd(pph3)4、pd(dppf)cl2、pd(pph3)2cl2、pd(cy3)2cl2、pd(oac)2 等催化剂催化下,na2co3、k2co3、cs2co3、csf等碱作用下,80-160℃温度下反应8-16 小时,制得关键中间体
ⅴ
,关键中间体
ⅴ
继续与化合物vi在dmf、dmso、ch3cn、thf 溶剂中,与k2co3、cs2co3、csf、nah在0-120℃温度下,发生取代反应,4-16小时后制得化合物i。
[0113]
方法二:
[0114][0115]
化合物ii和vi在dmf、dmso、ch3cn、thf溶剂中,与k2co3、cs2co3、csf、 nah在0-120℃温度下,发生取代反应,4-16小时后制得化合物vii,化合物vii继续在 na2co3、k2co3、cs2co3、csf等碱作用下与有机硼酸(ⅲ)或有机硼酸酯(ⅳ)在 1,4-dioxane、dmf、dmso、dme、1,4-dioxane/h2o等溶剂中,pd(pph3)4、pd(dppf)cl2、pd(pph3)2cl2、pd(cy3)2cl2、pd(oac)2等催化剂催化下,80-160℃温度下反应8-16小时,制得化合物i。
[0116]
其中,x为br或cl,z为c或n,y为o或s,虚线代表z相连的c形成单键或双键;
[0117]
进一步的,本发明涉及式ⅰ、ⅰ'、ⅰ"的化合物的用途,在制备治疗或预防疼痛药物中的用途。
具体实施方式
[0118]
以下将结合实施例和实验例对本发明作进一步的详细描述,本发明的实施例和实验例仅用于说明本发明的技术方案,并非对本发明的限制,凡依照本发明公开的内容所作的任何本领域的等同置换,均属于本发明的保护范围。
[0119]
化合物的结构是核磁共振(1h nmr)或液质联用(lc-ms)来确定的。
[0120]
液质联用仪(lc-ms)为安捷伦g6120b(与液相agilent 1260配用);核磁共振仪(1h nmr) 为bruker avance-400或bruker avance-800,核磁共振(1h nmr)位移(δ)以百万分之一(ppm)的单位给出,测定溶剂为cdcl3,内标为四甲基硅烷(tms),化学位移是以10-6 (ppm)作为单位给出。
[0121]
本发明的术语“室温”是指温度处于10~25℃之间。
[0122]
实施例1:1-(2-吗啉代乙基)-7-(萘-2-基)喹啉-2(1h)-酮的制备
[0123][0124]
步骤一,室温下,将化合物iia(2.24g,10mmol),化合物ⅵa(2.24g,15mmol),cs2co
3 (9.75g,30mmol)加入到dmf(30ml)中,加热到100℃,反应6h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.87g黄色油状产物。收率:85%。
[0125]1h nmr(400mhz,cdcl3)δ7.68
–
7.60(m,2h),7.42(d,j=8.3hz,1h),7.34(dd, j=8.3,1.6hz,1h),6.70(d,j=9.4hz,1h),4.47
–
4.30(m,2h),3.80
–
3.67(m,4h), 2.73
–
2.53(m,6h)。
[0126]
步骤二,室温下,将化合物ⅶa(2.87g,8.5mmol),化合物萘-2-基硼酸(1.76g,10.2 mmol),csf(1.55g,10.2mmol)加入到1,4-dioxane/h2o(30ml,v/v=5/1)中,氮气置换后,再加入pd(dppf)cl2(0.62g,0.85mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1过柱,旋干,真空干燥得到2.62g黄色油状产物:1-(2
‑ꢀ
吗啉代乙基)-7-(萘-2-基)喹啉-2(1h)-酮。收率:80%,纯度为99.53%。
[0127]
esi-ms:m/z=385.2(m+h)
+
。
[0128]1h nmr(400mhz,cdcl3)δ8.06(s,1h),7.98
–
7.83(m,5h),7.78
–
7.71(m,2h), 7.56
–
7.46(m,3h),6.74(d,j=9.4hz,1h),4.54
–
4.41(m,2h),3.80
–
3.68(m,4h), 2.76
–
2.56(m,6h)。
[0129]
实施例2:1-(2-吗啉代乙基)-7-(萘-2-基)喹喔啉-2(1h)-酮的制备
[0130][0131]
步骤一,室温下,将化合物iia(2.25g,10mmol),化合物ⅵa(2.24g,15mmol),nah(1.2g,30mmol)加入到dmso(30ml)中,加热到100℃,反应6h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.74g黄色油状产物。收率:81%。esi-ms:m/z=339.2(m+h) +
。
[0132]
步骤二,室温下,将化合物ⅶb(2.74g,8.1mmol),化合物萘-2-基硼酸(1.67g,9.72 mmol),cs2co3(3.17g,9.72mmol)加入到dmf(30ml)中,氮气置换后,再加入pd(dppf)cl
2 (0.62g,0.85mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚: 乙酸乙酯=2:1过柱,旋干,真空干燥得到2.56g黄色油状产物:1-(2-吗啉代乙基)-7-(萘
ꢀ‑
2-基)喹喔啉-2(1h)-酮。收率:82.0%,纯度为99.42%。
[0133]
esi-ms:m/z=386.2(m+h)
+
。
[0134]1h nmr(400mhz,cdcl3)δ8.40(d,j=76.1hz,1h),8.21
–
8.06(m,2h),8.02
–
7.64 (m,6h),7.62
–
7.47(m,2h),4.71
–
4.37(m,2h),3.80
–
3.63(m,4h),2.97
–
2.70(m, 2h),2.62
–
2.51(m,4h)。
[0135]
实施例3:7-溴-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0136][0137]
步骤一,室温下,将化合物iia(2.26g,10mmol),化合物ⅵa(2.24g,15mmol),csf (4.56g,30mmol)加入到thf(50ml)中,加热到100℃,反应6h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.92g黄色油状产物。收率:86%,纯度为99.42%。
[0138]
esi-ms:m/z=340.2(m+h)
+
。
[0139]1h nmr(400mhz,cdcl3)δ7.32(d,j=1.7hz,1h),7.13(dd,j=7.9,1.8hz,1h), 7.03(d,j=8.0hz,1h),4.03(t,j=6.9hz,2h),3.77
–
3.65(m,4h),2.91
–
2.78(m,2h), 2.69
–
2.46(m,8h)。
[0140]
实施例4:7-(2-氯苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0141][0142]
步骤一,以实施例3的方法,制备7-溴-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)
‑ꢀ
酮(ⅶc);
[0143]
步骤二,室温下,将化合物ⅶc(2.88g,8.5mmol),化合物2-氯苯硼酸(1.59g,10.2mmol), cs2co3(3.32g,10.2mmol)加入到dmso(30ml)中,氮气置换后,再加入pd(dppf)cl2(0.62 g,0.85mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1过柱,旋干,真空干燥得到2.57g白色油状:7-(2-氯苯基)-1-(2-吗啉代乙基)-3,4
‑ꢀ
二氢喹啉-2(1h)-酮,收率:81.5%,纯度为99.32%。
[0144]
esi-ms:m/z=371.5(m+h)
+
。
[0145]1h nmr(400mhz,cdcl3)δ7.52
–
7.46(m,1h),7.37
–
7.29(m,3h),7.24(d,j=7.6 hz,1h),7.20(d,j=1.3hz,1h),7.06(dd,j=7.6,1.5hz,1h),4.14
–
4.07(m,2h), 3.72
–
3.61(m,4h),3.01
–
2.89(m,2h),2.73
–
2.59(m,4h),2.59
–
2.48(m,4h)。
[0146]
实施例5:1-(2-吗啉代乙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)-酮的制备
[0147]
[0148]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的2-萘硼酸,得到白色油状的标题化合物,收率:81.2%,纯度为99.60%。
[0149]
esi-ms:m/z=387.2(m+h)
+
。
[0150]1h nmr(400mhz,cdcl3)δ8.01(d,j=1.1hz,1h),7.96
–
7.85(m,3h),7.71(dd, j=8.5,1.8hz,1h),7.56
–
7.47(m,2h),7.45(d,j=1.3hz,1h),7.36(dd,j=7.7,1.5 hz,1h),7.28(d,j=7.7hz,1h),4.24
–
4.15(m,2h),3.75
–
3.64(m,4h),3.01
–
2.90(m, 2h),2.75
–
2.62(m,4h),2.61
–
2.45(m,4h)。
[0151]
实施例6:7-(4-氟苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0152][0153]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-氟苯硼酸,得到白色油状的标题化合物,收率:83.1%,纯度为99.51%。
[0154]
esi-ms:m/z=455.2(m+h)
+
。
[0155]1h nmr(400mhz,cdcl3)δ7.55
–
7.48(m,2h),7.28
–
7.21(m,2h),7.20
–
7.10(m,3h),4.19
–
4.13(m,2h),3.74
–
3.63(m,4h),2.99
–
2.89(m,2h),2.76
–
2.59(m,4h), 2.59
–
2.47(m,4h)。
[0156]
实施例7:7-(4-氟-2-甲基苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0157][0158]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-氟-2-甲基苯硼酸,得到白色油状的标题化合物,收率:82.2%,纯度为99.75%。
[0159]
esi-ms:m/z=369.2(m+h)
+
。
[0160]1h nmr(400mhz,cdcl3)δ7.23
–
7.14(m,2h),7.02
–
6.89(m,4h),4.12
–
4.06(m, 2h),3.72
–
3.60(m,4h),3.01
–
2.87(m,2h),2.73
–
2.65(m,2h),2.62
–
2.55(m,2h), 2.56
–
2.46(m,4h),2.27(s,3h)。
[0161]
实施例8:1-(2-吗啉代乙基)-7-苯基-3,4-二氢喹啉-2(1h)-酮的制备
(3,4-二氟苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0176][0177]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的3,4-二氟苯硼酸,得到白色油状的标题化合物,收率:78.2%,纯度为99.32%。
[0178]
esi-ms:m/z=373.2(m+h)
+
。
[0179]1h nmr(400mhz,cdcl3)δ7.40
–
7.32(m,1h),7.30
–
7.20(m,4h),7.16(dd,j=7.7,1.6hz,1h),4.20
–
4.12(m,2h),3.76
–
3.62(m,4h),2.99
–
2.89(m,2h),2.73
–
2.60(m, 4h),2.59
–
2.49(m,4h)。
[0180]
实施例12:1-(2-吗啉代乙基)-7-(4-(三氟甲氧基)苯基)-3,4-二氢喹啉-2(1h)-酮的制备
[0181][0182]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-(三氟甲氧基)苯硼酸,得到白色油状的标题化合物,收率:83.2%,纯度为99.64%。
[0183]
esi-ms:m/z=421.2(m+h)
+
。
[0184]1h nmr(400mhz,cdcl3)δ7.60
–
7.54(m,2h),7.31(d,j=8.1hz,2h),7.26
–
7.22 (m,2h),7.19(dd,j=7.7,1.5hz,1h),4.21
–
4.11(m,2h),3.75
–
3.64(m,4h),2.99
–
2.88(m,2h),2.74
–
2.59(m,4h),2.60
–
2.47(m,4h)。
[0185]
实施例13:7-(4-甲氧基苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0186][0187]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-甲氧基苯硼酸,得到白色油状的标题化合物,收率:82.1%,纯度为99.09%。
[0188]
esi-ms:m/z=367.2(m+h)
+
。
[0189]1h nmr(400mhz,cdcl3)δ7.56
–
7.45(m,2h),7.28
–
7.27(m,1h),7.25
–
7.14(m, 2h),
7.04
–
6.95(m,2h),4.20
–
4.09(m,2h),3.86(s,3h),3.75
–
3.65(m,4h),2.92(dd, j=8.5,6.2hz,2h),2.73
–
2.61(m,4h),2.60
–
2.48(m,4h)。
[0190]
实施例14:7-(2-甲氧基苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0191][0192]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的2-甲氧基苯硼酸,得到白色油状的标题化合物,收率:81.2%,纯度为99.39%。
[0193]
esi-ms:m/z=367.2(m+h)
+
。
[0194]1h nmr(400mhz,cdcl3)δ7.38
–
7.27(m,3h),7.23
–
7.13(m,2h),7.08
–
6.98(m, 2h),4.19
–
4.05(m,2h),3.83(s,3h),3.72
–
3.61(m,4h),2.97
–
2.87(m,2h),2.71
–
2.61 (m,4h),2.60
–
2.47(m,4h)。
[0195]
实施例15:1-(2-吗啉代乙基)-7-(2-(三氟甲氧基)苯基)-3,4-二氢喹啉-2(1h)-酮的制备
[0196][0197]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的2-(三氟甲氧基)苯硼酸,得到白色油状的标题化合物,收率:80.7%,纯度为99.10%。
[0198]
esi-ms:m/z=421.2(m+h)
+
。
[0199]1h nmr(400mhz,cdcl3)δ7.44
–
7.38(m,2h),7.32(d,j=0.8hz,1h),7.23
–
7.12 (m,4h),4.13
–
4.02(m,2h),3.70
–
3.57(m,4h),2.92
–
2.81(m,2h),2.64
–
2.54(m, 4h),2.50
–
2.44(m,4h)。
[0200]
实施例16:7-(2-(甲硫基)苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0201][0202]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的2-甲硫基苯硼
酸,得到白色油状的标题化合物,收率:80.7%,纯度为99.28%。
[0203]
esi-ms:m/z=383.2(m+h)
+
。
[0204]1h nmr(400mhz,cdcl3)δ7.39
–
7.33(m,1h),7.31
–
7.27(m,1h),7.25
–
7.20(m, 3h),7.18(d,j=1.2hz,1h),7.04(dd,j=7.6,1.4hz,1h),4.15
–
4.05(m,2h),3.71
–ꢀ
3.59(m,4h),2.99
–
2.88(m,2h),2.74
–
2.59(m,4h),2.58
–
2.48(m,4h),2.39(s,3h)。
[0205]
实施例17:1-(2-吗啉代乙基)-7-(吡啶-3-基)-3,4-二氢喹啉-2(1h)-酮的制备
[0206][0207]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的3-吡啶硼酸,得到白色油状的标题化合物,收率:82.5%,纯度为99.29%。
[0208]
esi-ms:m/z=338.2(m+h)
+
。
[0209]1h nmr(400mhz,cdcl3)δ8.83(d,j=1.9hz,1h),8.62(dd,j=4.8,1.5hz,1h), 7.89
–
7.82(m,1h),7.40(dd,j=7.9,4.8hz,1h),7.33
–
7.26(m,2h),7.25
–
7.18(m, 1h),4.21
–
4.10(m,2h),3.76
–
3.62(m,4h),3.04
–
2.89(m,2h),2.75
–
2.60(m,4h),2.60
–
2.48(m,4h)。
[0210]
实施例18:7-(3,4-二氯苯基)-1-[2-(哌啶-1-基)乙基]-3,4-二氢喹啉-2(1h)-酮的制备
[0211][0212]
步骤一,室温下,将化合物ⅱc(2.26g,10mmol),3,4-二氯苯硼酸(2.29g,12mmol), cs2co3(3.91g,12mmol)加入到1,4-dioxane(30ml)中,氮气置换后,再加入pd(ph3p)
4 (1.16g,1mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.33g黄色油状产物。收率:80.1%。esi-ms:m/z= 292.2(m+h)
+
。
[0213]
步骤二,0℃,将化合物
ⅴ
a(2.34g,9.31mmol),化合物ⅵb(2.06g,13.96mmol),加入到无水thf(30ml)中,分批加入nah(0.56g,13.96mmol),升到室温,反应6h,原料反应完全。加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1过柱,旋干,真空干燥得到2.87g黄色油状产物。收率:76.3%,纯度为99.90%。
[0214]
esi-ms:m/z=404.2(m+h)
+
。
[0215]1h nmr(400mhz,cdcl3)δ7.72(d,j=1.9hz,1h),7.54
–
7.48(m,2h),7.38(d, j=1.2hz,1h),7.25
–
7.14(m,2h),4.34
–
4.19(m,2h),3.01
–
2.88(m,2h),2.88
–
2.71 (m,6h),2.72
–
2.61(m,2h)。
[0216]
实施例19:7-[3-(萘-2-基)苯基]-1-[2-(哌啶-1-基)乙基]-3,4-二氢喹啉-2(1h)-酮的制备
[0217][0218]
按实施例18的制备方法,将步骤一中的3,4-二氯苯硼酸换成等摩尔量的2-萘硼酸,得到淡褐色固体的标题化合物,收率:81.2%,纯度为99.63%。
[0219]
esi-ms:m/z=385.2(m+h)
+
。
[0220]1h nmr(400mhz,cdcl3)δ8.05(s,1h),7.96
–
7.84(m,3h),7.75(dd,j=8.5,1.8hz, 1h),7.56
–
7.45(m,3h),7.39
–
7.33(m,1h),7.30
–
7.26(m,1h),4.28
–
4.16(m,2h), 3.01
–
2.88(m,2h),2.76
–
2.47(m,8h),1.70
–
1.53(m,4h),1.51
–
1.36(m,2h)。
[0221]
实施例20:7-[2-(甲硫基)苯基]-1-[2-(哌啶-1-基)乙基]-3,4-二氢喹啉-2(1h)-酮的制备
[0222][0223]
按实施例18的制备方法,将步骤一中的2-氯苯硼酸换成等摩尔量的2-(甲硫基)苯硼酸,得到淡黄色油状的标题化合物,收率:82.9%,纯度为99.36%。
[0224]
esi-ms:m/z=381.2(m+h)
+
。
[0225]1h nmr(400mhz,cdcl3)δ7.38
–
7.32(m,1h),7.30
–
7.27(m,1h),7.24
–
7.17(m, 4h),7.04(dd,j=7.6,1.5hz,1h),4.21
–
4.05(m,2h),3.00
–
2.87(m,2h),2.75
–
2.44 (m,8h),2.39(s,3h),1.63
–
1.50(m,4h),1.48
–
1.34(m,2h)。
[0226]
实施例21:7-(二苯并[b,d]噻吩-4-基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0227]
[0228]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的二苯并噻吩-4-硼酸,得到淡黄色油状的标题化合物,收率:76.2%,纯度为99.23%。
[0229]
esi-ms:m/z=443.2(m+h)
+
。
[0230]1h nmr(400mhz,cdcl3)δ8.24
–
8.11(m,2h),7.86
–
7.78(m,1h),7.57(t,j=7.6hz, 1h),7.54
–
7.43(m,4h),7.40
–
7.29(m,2h),4.24
–
4.12(m,2h),3.72
–
3.55(m,4h), 3.07
–
2.92(m,2h),2.80
–
2.68(m,4h),2.68
–
2.51(m,4h)。
[0231]
实施例22:4-[1-(2-吗啉代乙基)-2-氧代-1,2,3,4-四氢喹啉-7-基]苄腈的制备
[0232][0233]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-氰基苯硼酸,得到淡黄色油状的标题化合物,收率:80.2%,纯度为99.30%。
[0234]
esi-ms:m/z=362.2(m+h)
+
。
[0235]1h nmr(400mhz,cdcl3)δ7.78
–
7.72(m,2h),7.72
–
7.64(m,2h),7.35
–
7.18(m, 3h),4.24
–
4.13(m,2h),3.80
–
3.62(m,4h),3.01
–
2.88(m,2h),2.74
–
2.52(m,8h)。
[0236]
实施例23:1'-(2-吗啉代乙基)-3',4'-二氢-[-4,7'-双喹啉]-2'(1'h)-酮的制备
[0237][0238]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-喹啉硼酸,得到淡黄色油状的标题化合物,收率:78.9%,纯度为98.71%。
[0239]
esi-ms:m/z=388.2(m+h)
+
。
[0240]1h nmr(400mhz,cdcl3)δ8.99(d,j=4.4hz,1h),8.23(d,j=8.2hz,1h),7.93(dd, j=8.4,0.7hz,1h),7.84
–
7.71(m,1h),7.59
–
7.50(m,1h),7.38(d,j=4.5hz,1h), 7.34(d,j=7.6hz,1h),7.23(d,j=1.2hz,1h),7.16(dd,j=7.6,1.4hz,1h),4.14
–
4.10 (m,2h),3.68
–
3.56(m,4h),3.08
–
2.97(m,2h),2.80
–
2.71(m,2h),2.70
–
2.61(m, 2h),2.62
–
2.49(m,4h)。
[0241]
实施例24:1-(3-吗啉代丙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)-酮的制备
[0242]
步骤一,室温下,将化合物ⅱc(2.26g,10mmol),萘-2-硼酸酯(3.05g,12mmol), csf(1.82g,12mmol)加入到1,4-dioxane(30ml)中,氮气置换后,再加入pd(dppf)cl2(0.73 g,1mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.18g黄色油状产物
ⅴ
b。收率:79.6%。esi-ms:m/z=274.2(m+h) +
。
[0243]
步骤二,0℃,将化合物
ⅴ
b(2.18g,8.0mmol),化合物ⅵc(2.16g,11.7mmol),加入到无水dmf(30ml)中,加入csf(1.78g,11.7mmol),升到室温,反应6h,原料反应完全。加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1过柱,旋干,真空干燥得到2.43g黄色油状产物:1-(3-吗啉代乙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)
ꢀ‑
酮。收率:76.4%,纯度为98.11%。
[0244]
esi-ms:m/z=401.2(m+h)
+
。
[0245]1h nmr(400mhz,cdcl3)δ8.00(s,1h),7.95
–
7.85(m,3h),7.70(dd,j=8.5,1.8hz, 1h),7.56
–
7.47(m,2h),7.37
–
7.32(m,2h),7.30
–
7.25(m,1h),4.13
–
4.05(m,2h), 3.71
–
3.62(m,4h),3.00
–
2.89(m,2h),2.75
–
2.65(m,2h),2.58
–
2.43(m,6h),2.01
–ꢀ
1.92(m,2h)。
[0246]
实施例25:1-[2-(氮杂环庚烷-1-基)乙基]-7-(2-甲硫苯基)-3,4-二氢喹啉-2(1h)-酮的制备
[0247][0248]
步骤一,室温下,将化合物ⅱc(2.26g,10mmol),2-甲硫基苯硼酸酯(3.00g,12mmol), k2co3(1.66g,12mmol)加入到dmso(30ml)中,氮气置换后,再加入pd(dppf)cl2(0.73 g,1mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.10g黄色油状产物
ⅴ
c,收率:78%。esi-ms:m/z= 270.2(m+h)
+
。
[0249]
步骤二,在0℃时,将化合物
ⅴ
c(2.10g,7.8mmol),化合物ⅵd(1.89g,11.7mmol),加入到无水ch3cn(50ml)中,加入cs2co3(3.89g,11.7mmol),升到室温,反应6h,原料反应完全。加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1 过柱,旋干,真空干燥得到2.33g黄色油状产物:1-[2-(氮杂环庚烷-1-基)乙基]-7-(2-甲硫苯基)-3,4-二氢喹啉-2(1h)-酮。收率:75%,纯度为98.11%。
[0250]
esi-ms:m/z=395.2(m+h)
+
。
[0251]1h nmr(400mhz,cdcl3)δ7.38
–
7.31(m,1h),7.31
–
7.27(m,1h),7.25
–
7.17(m, 4h),
7.06(dd,j=7.5,1.4hz,1h),4.30
–
4.18(m,2h),3.17
–
3.00(m,6h),2.99
–
2.85 (m,2h),2.75
–
2.63(m,2h),2.39(s,3h),1.93
–
1.70(m,8h)。
[0252]
实施例26:1-[2-(氮杂环庚烷-1-基)乙基]-6-甲氧基-3,4-二氢喹啉-2(1h)-酮的制备
[0253][0254]
在0℃时,将化合物ⅱd(1.77g,10mmol),化合物ⅵa(2.42g,15mmol),加入到无水dmso(30ml)中,加入k2co3(2.07g,15mmol),升到室温,反应6h,原料反应完全。加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1过柱,旋干,真空干燥得到2.42g黄色油状产物ⅶd。收率:80.2%,纯度为98.11%。
[0255]
esi-ms:m/z=303.2(m+h)
+
。
[0256]1h nmr(400mhz,cdcl3)δ7.04(d,j=8.8hz,1h),6.81
–
6.68(m,2h),4.15
–
4.01(m, 2h),3.79(s,3h),2.99
–
2.76(m,4h),2.67
–
2.42(m,8h),1.71
–
1.54(m,4h),1.52
–
1.40 (m,2h)。
[0257]
实施例27:7-(3,4-二氯苯基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-硫酮的制备
[0258][0259]
步骤一,按实施例3的制备方法制得中间体7-溴-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)
ꢀ‑
酮(ⅶc)
[0260]
步骤二,将化合物7-溴-1-(2-吗啉代吗啡酮)-3,4-二氢喹啉-2(1h)-酮(2.72g,8mmol) 溶于甲苯(40ml)中,加入劳森试剂(1.64g,2mmol),此悬浮液加热120℃,反应4小时,然后将体系冷却至10℃并有固体板出,此固体用布氏漏斗过滤收集,亲用少量二氯甲烷洗涤,经干燥后得到7-溴-1-(2-吗啉代吗啡酮)-3,4-二氢喹啉-2(1h)-硫酮(2.08g),收率73%。
[0261]
步骤三,室温下,将化合物
ⅸ
c(7-溴-1-(2-吗啉代吗啡酮)-3,4-二氢喹啉-2(1h)-硫酮)(2.08g,5.85mmol),化合物3,4-二氯苯基硼酸(1.42g,7.44mmol),csf(1.13g, 10.2mmol)加入到dme(50ml)中,氮气置换后,再加入pd(pph3)2cl2(0.44g,0.85mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1过柱,旋干,真空干燥得到1.90g黄色油状产物。收率:79.3%,纯度为98.71%。
[0262]
esi-ms:m/z=387.3(m+h)
+
。
[0263]1h nmr(800mhz,cdcl3)δ8.07(s,1h),7.90(d,j=8.6hz,1h),7.82(s,1h),7.79 (d,j=8.3hz,1h),7.62(dd,j=8.3,1.7hz,1h),7.23(d,j=8.6hz,1h),3.84
–
3.68(m, 6h),3.54(brs,2h),2.88
–
2.52(m,8h)。
[0264]
实施例28:7-(2-氯苯基)-1-[2-(二甲基氨基)乙基]-3,4-二氢喹啉-2(1h)-酮的制备
[0265]
步骤一,室温下,将化合物iic(2.26g,10mmol),化合物ⅵe(1.61g,15mmol), k2co3(4.14g,30mmol)加入到ch3cn(50ml)中,加热到100℃,反应6h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚: 乙酸乙酯=3:1过柱,旋干,真空干燥得到2.49g黄色油状产物ⅶe。收率:83.8%。esi-ms:m/z =297.2(m+h)
+
。
[0266]
步骤二,室温下,将化合物ⅶe(2.49g,8.4mmol),化合物2-氯苯硼酸(1.58g,10.1mmol), csf(1.53g,10.2mmol)加入到dmso(30ml)中,氮气置换后,再加入pd(pph3)4(0.97g,0.84mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=2:1 过柱,旋干,真空干燥得到2.25g黄色油状产物7-(2-氯苯基)-1-[2-(二甲基氨基)乙基]-3,4
‑ꢀ
二氢喹啉-2(1h)-酮。收率:81.3%,纯度为98.85%。
[0267]
esi-ms:m/z=329.2(m+h)
+
。
[0268]1h nmr(400mhz,cdcl3)δ7.52
–
7.46(m,1h),7.37
–
7.29(m,3h),7.24(d,j=7.6 hz,1h),7.20(d,j=1.3hz,1h),7.06(dd,j=7.6,1.5hz,1h),4.13
–
4.08(m,2h), 3.00
–
2.90(m,2h),2.74
–
2.60(m,4h),2.33(s,6h)。
[0269]
实施例29:6-(3,4-二氯苯基)-1-(2-(哌啶-1-基)乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0270][0271]
步骤一,室温下,将6-溴-3,4-二氢喹啉-2(1h)-酮(2.26g,10mmol),3,4-二氯苯硼酸(2.29g,12mmol),cs2co3(3.91g,12mmol)加入到1,4-dioxane(30ml)中,氮气置换后,再加入pd(ph3p)4(1.16g,1mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3) 萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油
1h),3.78
–
3.63(m,6h),3.48
–
3.38(m,2h),3.33-3.23(m,2h),2.75-2.67(m,2h),2.58
ꢀ–
2.45(m,6h),1.96
–
1.88(m,2h),1.84
–
1.71(m,2h)。
[0284]
实施例31:7-(3-氟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0285][0286]
步骤一,室温下,将实施例化合物30(0.9g,2.8mmol)溶解在无水二氯甲烷(20ml) 中,氮气置换保护后,冷却到-78℃,滴加二乙胺基三氟化硫(0.68g,4.2mmol)。滴加完毕,反应液慢慢升温到室温,搅拌16h。反应液冷却到0℃,用饱和nahco3溶液淬灭反应,加入h2o(30ml),用二氯甲烷(50ml*3)萃取三次,有机相依次用水,饱和nahco3溶液,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂甲醇:二氯甲烷=20:1 过柱,旋干,真空干燥得到0.4g黄色油状标题化合物7-(3-氟丙基)-1-(2-吗啉代乙基)-3,4
‑ꢀ
二氢喹啉-2(1h)-酮。收率:44.6%,纯度99.35%。
[0287]
esi-ms:m/z=321.2(m+h)
+
。
[0288]1h nmr(800mhz,cdcl3)δ7.09(d,j=7.5hz,1h),6.92(s,1h),6.86
–
6.83(m,1h),4.51
–
4.43(m,2h),4.13
–
4.04(m,2h),3.71(t,j=4.5hz,4h),2.88
–
2.83(m,2h),2.77
ꢀ–
2.70(m,2h),2.66
–
2.62(m,2h),2.60
–
2.57(m,2h),2.55(brs,4h),2.07
–
1.93(m, 2h)。
[0289]
实施例32:6-(3-羟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0290][0291]
步骤一,室温下,将化合物ⅱd(2.26g,10mmol),化合物ⅵa(2.24g,15mmol), cs2co3(9.75g,30mmol)加入到dmf(30ml)中,加热到100℃,反应6h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂石油醚:乙酸乙酯=3:1过柱,旋干,真空干燥得到2.86g黄色油状化合物ⅶf。收率:85%。
[0292]
步骤二,室温下,将化合物ⅶf(3g,8.8mmol),烯丙硼酸(0.91g,10.6mmol),csf (4.01g,26.4mmol)加入到1,4-dioxane/h2o(60ml,v/v=5/1)中,氮气置换后,再加入 pd(dppf)cl2(0.64g,0.88mmol),再次氮气置换三次,氮气保护下加热到100℃,反应16h,原料反应完全。将反应液冷却到室温,加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂甲醇:二氯甲烷=20:1过柱,旋干,真空干燥得到2.4g黄色油状标题化合物6-烯丙基-1-(2
‑ꢀ
吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮。收率:90.9%,纯度99.23%。
[0293]
esi-ms:m/z=301.2(m+h)
+
。
[0294]1h nmr(800mhz,cdcl3)δ7.06(dd,j=8.3,1.5hz,1h),7.01
–
6.96(m,2h),5.97
ꢀ–
5.91(m,1h),5.12
–
5.05(m,2h),4.12
–
4.03(m,2h),3.70(t,j=4.5hz,4h),3.34(d, j=6.7hz,2h),2.92
–
2.82(m,2h),2.64
–
2.61(m,2h),2.59
–
2.56(m,2h),2.54(brs, 4h)。
[0295]
步骤三,室温下,将
ⅺ
b(1g,3.3mmol)溶解在无水thf(20ml)中,氮气置换保护后,冷却到-40℃,滴加1m bh3.thf(10ml)。滴加完毕,反应液慢慢升温到室温,反应2h。反应液再冷却到0℃,滴加h2o(10ml)淬灭多余的bh3.thf,再滴加0.5m naoh(10ml)和 35%h2o2(2ml),并在0℃下反应1h。加入h2o(30ml),用乙酸乙酯(50ml*3)萃取三次,有机相依次用水,饱和nahco3溶液,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂甲醇:二氯甲烷=20:1过柱,旋干,真空干燥得到0.34g黄色油状标题化合物6-(3-羟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮。收率:32.5%,纯度99.16%。
[0296]
esi-ms:m/z=319.2(m+h)
+
。
[0297]1h nmr(800mhz,cdcl3)δ7.07(dd,j=8.2,1.8hz,1h),7.01(s,1h),6.98(d,j= 8.3hz,1h),4.13
–
4.03(m,2h),3.75
–
3.65(m,6h),2.88
–
2.83(m,2h),2.69
–
2.65(m, 2h),2.64
–
2.61(m,2h),2.59
–
2.56(m,2h),2.54(brs,4h),1.91
–
1.85(m,2h)。
[0298]
实施例33:6-(3-氟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0299][0300]
步骤一,室温下,将实施例化合物32(0.9g,2.8mmol)溶解在无水二氯甲烷(20ml) 中,氮气置换保护后,冷却到-78℃,滴加dast(0.68g,4.2mmol)。滴加完毕,反应液慢慢升温到室温,搅拌16h。反应液冷却到0℃,用饱和nahco3溶液淬灭反应,加入h2o(30ml),用二氯甲烷(50ml*3)萃取三次,有机相依次用水,饱和nahco3溶液,饱和食盐水洗涤,有机相用无水硫酸钠干燥,过滤,滤液旋干,粗品用洗脱剂甲醇:二氯甲烷=20:1过柱,旋干,真空干燥得到0.43g黄色油状标题化合物6-(3-氟丙基)-1-(2-吗啉代乙基)-3,4-二氢喹啉
ꢀ‑
2(1h)-酮。收率:48.2%,纯度98.99%。
[0301]
esi-ms:m/z=321.2(m+h)
+
。
[0302]1h nmr(800mhz,cdcl3)δ7.11
–
7.06(m,1h),7.03
–
6.97(m,2h),4.50
–
4.41(m, 2h),4.08(t,j=7.3hz,2h),3.72(brs,4h),2.88-2.85(m,2h),2.72
–
2.67(m,2h),2.65
ꢀ–
2.49(m,8h),2.04-1.94(m,2h)。
[0303]
实施例34:7-([1,1'-联苯]-3-基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0304][0305]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的3-联苯硼酸,得
到淡黄色油状的标题化合物,收率:83.2%,纯度为99.77%。
[0306]
esi-ms:m/z=413.2(m+h)
+
。
[0307]1h nmr(800mhz,cdcl3)δ7.76(s,1h),7.63(d,j=7.6hz,2h),7.60(d,j=7.1hz, 1h),7.56
–
7.51(m,2h),7.47(t,j=7.6hz,2h),7.38(t,j=7.4hz,1h),7.36(s,1h), 7.29(d,j=7.7hz,1h),7.25(s,1h),4.16(t,j=7.2hz,2h),3.65(t,j=4.3hz,4h), 2.97
–
2.92(m,2h),2.71
–
2.67(m,2h),2.64(t,j=7.2hz,2h),2.55(brs,4h)。
[0308]
实施例35:7-([1,1'-联苯]-4-基)-1-(2-吗啉代乙基)-3,4-二氢喹啉-2(1h)-酮的制备
[0309][0310]
按实施例4的制备方法,将步骤二中的2-氯苯硼酸换成等摩尔量的4-联苯基硼酸,得到淡黄色油状的标题化合物,收率:81.1%,纯度为99.93%。
[0311]
esi-ms:m/z=413.2(m+h)
+
。
[0312]1h nmr(800mhz,cdcl3)δ7.69(d,j=8.2hz,2h),7.65(d,j=8.2hz,4h),7.47(t, j=7.7hz,2h),7.39
–
7.35(m,2h),7.28(dd,j=7.6,1.3hz,1h),7.25(d,1h),4.17(t, j=7.2hz,2h),3.70(t,j=4.5hz,4h),2.97
–
2.90(m,2h),2.71
–
2.63(m,4h),2.56(brs, 4h)。
[0313]
实施例36:1-(2-吗啉代乙基)-7-(萘-2-基)-3,4-二氢喹啉-2(1h)-硫酮的制备
[0314][0315]
按实施例27的制备方法,将步骤三中的3,4-二氯苯硼酸换成等摩尔量的2-萘硼酸,得到淡黄色油状的标题化合物,收率:80.1%,纯度为98.57%。
[0316]
esi-ms:m/z=403.2(m+h)
+
。
[0317]1h nmr(800mhz,cdcl3)δ8.39
–
8.18(m,2h),8.13
–
7.74(m,6h),7.66
–
7.47(m, 3h),5.41
–
5.28(m,1h),4.75(brs,2h),4.18
–
4.04(m,1h),3.93
–
3.42(m,6h),2.94
–ꢀ
2.48(m,4h),2.12
–
1.95(m,2h)。
[0318]
制备例1:4-{2
–
[[5-甲基-1-(萘-2-基)-1h-吡唑-3-基]氧基]乙基}吗啉的制备
[0319][0320]
步骤一,将化合物1h(10g,61mmol)溶解于100ml甲苯中,再向反应液中加入100ml 乙酸乙酯。反应液在冰浴下降温到0℃,醋酸酐(7.74g,75mmol)缓慢滴加入反应液中,滴完后自然升温至室温,反应过夜。通过tlc(二氯甲烷:甲醇=10:1)检测原料反应完。向反应液中加入100ml水,用饱和na2co3溶液调节ph=9-10,反应液用乙酸乙酯(100ml* 3)萃取,有机相用饱和nahco3溶液,水,饱和食盐水分别洗涤,用无水硫酸钠干燥。有机相过滤,滤液浓缩得粗品,粗品用柱层析(石油醚:乙酸乙酯=5:1~2:1)纯化得化合物2h(7.6 g,淡黄色油状物)。收率:62.3%。esi-ms:m/z=201.2(m+h)
+
。
[0321]
步骤二,将化合物2h(6g,30mmol),乙酰乙酸乙酯(4.85g,45mmol),在氮气保护下,缓慢加入到三溴化磷(10ml)中,反应液升温至50℃,保温反应2小时。通过tlc(石油醚:乙酸乙酯=1:1)检测原料反应完。将反应液降温至室温,倒入100ml二氯甲烷中,加入50ml冰水,用二氯甲烷(100ml*3)萃取,有机相用饱和nahco3溶液,水,饱和食盐水分别洗涤,用无水硫酸钠干燥。有机相过滤,滤液浓缩得粗品,粗品用柱层析(石油醚:乙酸乙酯=5:1~2:1)纯化得化合物3h(2.2g,白色固体)。收率:32.7%。esi-ms:m/z=225.2 (m+h)
+
。
[0322]
步骤三,在氮气保护下,将化合物3h(2g,8.9mmol),n-(2-氯乙基)吗啉盐酸盐(1.6g, 10.7mmol)加入到10ml dmf中,在冰浴下分批加入nah(0.72g,17.8mmol)。反应液升温至60℃反应4小时。通过tlc(石油醚:乙酸乙酯=1:1)检测原料反应完。将反应液降温至室温,倒入100ml乙酸乙酯中,加入50ml冰水,用乙酸乙酯(100ml*3)萃取,有机相用水,饱和食盐水分别洗涤,用无水硫酸钠干燥。有机相过滤,滤液浓缩得粗品,粗品用柱层析(石油醚:乙酸乙酯=5:1~1:1)纯化得化合物4h(2.3g,白色固体)。收率:76.6%。esi-ms: m/z=338.2(m+h)
+
。
[0323]
步骤四,将化合物4h(2.3g,6.8mmol)溶解于3ml无水乙酸乙酯中,在冰浴下滴加乙酸乙酯/hcl(2.5n,3ml),滴完自然恢复至室温,反应过夜。将反应液过滤,滤渣用无水乙酸乙酯洗涤三次,固体干燥制得制备例1标题化合物(2.1g,白色固体)收率:82.7%.纯度为 98.47%。
[0324]1h nmr(400mhz,cdcl3)δ7.95
–
7.78(m,4h),7.60(dd,j=8.7,2.1hz,1h),7.55
ꢀ–
7.47(m,2h),5.73(s,1h),4.38(t,j=5.6hz,2h),3.79
–
3.71(m,4h),2.81(t,j=5.6 hz,2h),2.65
–
2.50(m,4h),2.36(s,3h)。
[0325]
试验例1:小鼠醋酸扭体试验
[0326]
1.试验材料
[0327]
1.1试验试剂
空白组 30.2 制备例1 28.5 实施例4 1.3 实施例5 0.0 实施例6 19.1 实施例7 9.1 实施例8 15.3 实施例9 18.8 实施例10 1.9 实施例11 18.1 实施例12 25.6 实施例13 5.4 实施例14 0.2 实施例15 0.0 实施例16 1.4 实施例17 24.8 实施例18 12 实施例19 5.6 实施例20 4.4 实施例22 19.7 实施例23 3.8 实施例24 13.6 实施例25 12.5 实施例26 11.5 实施例28 20.8 实施例29 18.3 实施例36 15.3 [0351]
如上表所示,腹腔给予2.5mg/kg实施例化合物后,与空白组相比,实施例4、实施例5、实施例7、实施例10、实施例13、实施例14、实施例15、实施例16、实施例20、实施例24、实施例26,同样在小鼠醋酸扭体试验中均表现出强大的镇痛作用。制备例1在此条件进行的小鼠醋酸扭体试验中与空白组相比,没有表现出有统计学意义的镇痛活性。
[0352]
表1-4:腹腔注射1mg/kg实验例化合物时,小鼠的平均扭体次数
[0353]
[0354][0355]
如上表所示,腹腔给予1mg/kg实施例化合物后,实施例化合物5、10、15、16同样在小鼠醋酸扭体试验中表现出明显的镇痛作用。制备例1在此条件进行的小鼠醋酸扭体试验中与空白组相比,没有表现出有统计学意义的镇痛活性。
[0356]
表1-5:腹腔注射0.5mg/kg实验例化合物时,小鼠的平均扭体次数
[0357]
编号 小鼠的平均扭体次数 dmso 29.6 制备例1 31.2 实施例5 10.3 实施例10 15.2 实施例15 18.9 实施例16 20.1 [0358]
如上表所示,腹腔给予0.5mg/kg实施例化合物后,与空白组相比,实施例5、实施例10、实施例15、实施例16化合物,同样在小鼠醋酸扭体试验中表现出镇痛作用,特别是实施例5 所述化合物。制备例1在此条件进行的小鼠醋酸扭体试验中与空白组相比,没有表现出有统计学意义的镇痛活性。
[0359]
试验例2:对热传导引起小鼠拟痛反应的影响试验(热板法试验)
[0360]
1.试验材料
[0361]
1.1试验试剂
[0362]
试剂名称 批号 来源 kolliphor hs-15 48328768e0 巴斯夫应用化工有限公司生产 0.9%氯化钠注射液 w215080801 四川科伦药业股份有限公司生产 羧甲基纤维素 #g1226001 阿拉丁 高级碳素墨水 201604285 贵州博士化工有限公司 药用碳片 48160118 河北长天药业有限公司 [0363] 1.2试验仪器
[0364]
移液器、离心管、电子天平、注射器、小鼠固定器、智能热板仪、尺子、剪刀、镊子、灌胃针等。
[0365]
2.实验动物
[0366]
昆明小鼠,全雌小鼠,18-22g,由成都达硕实验动物有限公司提供。
[0367]
3.试验方法
[0368]
3.1供试品配制
[0369]
生理盐水中加入hcl调ph至4.5,以备后用。
[0370]
准确称取供试品即制备例1、实施例5、实施例10化合物各2mg,分别加入50%的hs-15 溶液1ml,混匀后再加入1ml的生理盐水盐酸溶液,充分溶解并混匀,即为1mg/ml的供试品溶液。
[0371]
3.2试验操作
[0372]
取km全雌小鼠90只,18-22g,在55
±
0.5℃的热板上测定基础痛阈值,选痛阈值在5-30s 的小鼠40只供实验用。筛选合格的40只小鼠在给药前测痛阈值2次,间隔5min,取其平均值为基础痛阈值。随机分为4组,即空白对照组,制备例1、实施例5、实施例10化合物组,每组10只,各组尾静脉给药(对照组给予等量的0.9%生理盐水),给药体积均为1ml/kg,于给药后30、60、120min测定小鼠的痛阈值,痛阈值超过60s者按60s计,统计比较各药物组与空白对照组痛阈的差异。
[0373]
4.试验结果与讨论
[0374]
表2-1,制备例1、实施例5、实施例10化合物对热板致小鼠痛阈值的影响
[0375][0376][0377]
注:“*”与空白组相比,p<0.05;“**”与空白组相比,p<0.01。
[0378]
从上表可见,实施例5、实施例10化合物在药后30min、60min均能提高小鼠的痛阈值,与空白组比较有统计学差异;制备例1化合物在药后30min、60min和120min对小鼠的痛阈值均没有明显的提高作用,与空白组没有统计学差异。
[0379]
试验例3:大鼠热辐射甩尾试验
[0380]
3.1试验材料
[0381]
3.1.1供试品基本信息
[0382]
化合物编号 性状性状 批号 来源 制备例1 白色油状 20180314 自制 实施例5 淡黄色油状 20180813 自制 实施例10 淡黄色油状 20180802 自制 [0383]
3.1.2试验试剂
[0384]
试剂名称 批号 来源 kolliphor hs-15 69889088q0 北京凤礼精求医药股份有限公司 0.9%氯化钠注射液 w214011102 四川科伦药业股份有限公司生产 [0385]
3.1.3试验仪器
[0386]
1ml注射器、计时器、微量移液器、电子天平、15ml离心管、50ml离心管、鼠尾光照测痛仪等。
[0387]
3.2实验动物
[0388]
sd大鼠,全雌性,体重160-180g,由成都达硕生物科技有限公司提供。
[0389]
3.3试验方法
[0390]
3.3.1供试品配制
[0391]
生理盐水中加入hcl调ph至4.5,以备后用。
[0392]
称取各化合物约20mg于50ml离心管中,加入生理盐水盐酸溶液配制成1mg/ml的供试品溶液,难溶解化合物可加热助溶。制备例1加入100μl的50%hs-15助溶后加入生理盐水配制成1mg/ml的供试品溶液。
[0393]
3.3.2给药及检测
[0394]
适应期过后,在给药前一天对大鼠进行痛阈的测定。选用尾部的下1/3处作为测痛点,测定3次,测试间隔时间不少于5min,取3次测定的平均值为每只大鼠的基础痛阈。以照射开始到甩尾反应的潜伏期(tail flick latency,tfl)作为痛阈,调节测痛仪功率在99%。将基础痛阈小于2s或大于10s的动物剔除。根据基础痛阈选取24只动物随机分成4组,每组6只:
[0395][0396]
根据上述表格进行给药,分别在给药后的30、60、120min测试给药后的痛阈。
[0397]
3.4试验结果与讨论
[0398]
按照下列公式计算最大镇痛效应百分率(mpe%)和痛阈提高率:
[0399]
最大镇痛效应百分率(mpe%)=(给药后tfl-基础tfl)/(15-基础tfl)
×
100%
[0400]
痛阈提高率(%)=(给药后tfl-基础tfl)/基础tfl
×
100%
[0401]
本次试验完成了3个化合物的大鼠热辐射甩尾试验。试验结果如下:
[0402]
表3-1:试验化合物热辐射甩尾试验结果
[0403]
[0404][0405]
注:“*”与生理盐水组相比,p<0.05;“**”与生理盐水组相比,p<0.01;
[0406]
由上表可见:
[0407]
①
在本试验条件下,给药剂量为4mg/kg时,大鼠给药后30min,实施例5(最大镇痛效应100%、痛阈提高率92.87%)、实施例10(最大镇痛效应73.65%、痛阈提高率82.12%)镇痛效果最好;制备例1未观察到镇痛作用;
[0408]
②
本试验条件下,给药剂量为4mg/kg时,大鼠给药后60min,实施例5(最大镇痛效应87.41%、痛阈提高率82.23%)仍有较好的镇痛作用;实施例10(最大镇痛效应31.54%、痛阈提高率 35.87%)镇痛效果明显降低;制备例1未观察到镇痛作用;
[0409]
③
本试验条件下,给药剂量为4mg/kg时,大鼠给药后120min,实施例5(最大镇痛效应59.85%、痛阈提高率52.02%)仍有较好的镇痛作用,但镇痛作用明显降低;实施例10(最大镇痛效应 10.81%、痛阈提高率16.99%)镇痛效果进一步明显降低,镇痛作用较弱;制备例1未观察到镇痛作用;
[0410]
因此,通过大鼠热辐射甩尾试验检测实施例5和10化合物的镇痛作用,结果显示:本试验条件下,实施例5化合物镇痛作用最好,在30min、60min、120min与生理盐水相比,均具有显著的统计学差异(p《0.01);实施例10化合物镇痛作用次之,在给药30min时有较好的镇痛作用,与生理盐水相比,具有显著的统计学差异(p《0.01),给药60min时,与生理盐水相比,有统计学差异(p《0.05),随后镇痛效果逐渐降低;制备例1化合物未观察到镇痛作用,在30min、60min、120min与生理盐水相比,无统计学差异(p》0.05),对大鼠痛阈无影响。
[0411]
综上,本发明提供了新型烷基氨类化合物,在小鼠醋酸扭体试验、对热传导引起小鼠拟痛反应的影响试验及大鼠热辐射甩尾试验中均显现出了优异的镇痛作用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1