一种副溶血弧菌基因高效敲除质粒的构建方法
1.本发明涉及一种副溶血弧菌基因高效敲除质粒的构建方法,属于分子生物学和生物技术领域。
背景技术:
2.副溶血弧菌传统敲除流程为:将同源臂连接到自杀质粒上,通过接合转导进入细胞并同源重组整合到基因组上。其次在10%蔗糖的环境中,整合到基因组的质粒上的sacb会分解蔗糖产生致死物质,由此迫使细胞发生第二次同源重组去掉质粒。第二次同源重组会出现两种情况,第一种为同源重组发生的位置与第一步不同,即第一步与第二步的同源重组分别发生在上游/下游同源臂上,这样在去掉质粒的同时,目的基因片段也会随之去除,敲除成功;第二种情况为同源重组发生的位置与第一步相同,即第一步与第二步的同源重组发生在同一个同源臂上,结果为整合在基因组上的质粒原封不动的被去除掉,敲除失败。随着目的基因在细胞中的重要性不同,敲除的难易程度也会随之改变。在某些基因的敲除过程中,第二步会花费大量的时间和资源。
3.并且,sacb基因容易发生突变,其反向筛选的假阳性率高,所以最后需要应对大量突变株进行鉴定,花费大量的时间和资源。因此,急需一种准确率高的副溶血弧菌的敲除方法。
技术实现要素:
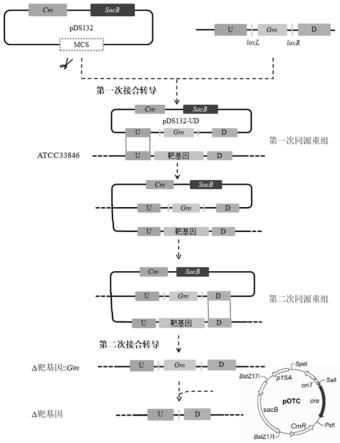
4.为了解决上述存在的技术问题,本发明优化了敲除过程,引入cre/loxp系统。在敲除同源臂之间加入了带有loxp位点的庆大霉素抗性片段,后该抗性片段通过cre酶去除。第二次同源重组时加入庆大霉素,只有正确进行同源重组的菌株可以存活于10%蔗糖庆大抗性平板上,以此提高敲除菌株的筛选效率。再进行第二次结合转导,将带有cre基因的potc质粒导入副溶血弧菌。cre基因在细胞中表达cre酶,识别loxp位点,并去除loxl与loxr之间的基因片段,即去除庆大抗性片段。最后在含有10%蔗糖的试管中去除potc质粒。
5.本发明提供了一种敲除质粒potc,所述敲除质粒potc包含复制子p15a、抗性基因cmr、转移元件orit和traj、启动子ptac、cre基因;所述敲除质粒potc的核苷酸序列如seq id no.1所示。
6.在本发明的一种实施方式中,所述敲除质粒potc中还含有sacb基因;所述sacb基因的核苷酸序列如seq id no.2所示。
7.本发明还提供了一种敲除副溶血弧菌中目的基因的方法,所述方法包括以下步骤:
8.(1)合成loxl-gm-loxr片段;
9.(2)将目的基因上下游同源臂同loxl-gm-loxr片段整合到pds132质粒上,得到重组质粒pds132-1;
10.(3)将重组质粒pds132-1导入大肠杆菌感受态中,得到重组大肠杆菌/pds132-1;
11.(4)将重组大肠杆菌/pds132-1同副溶血弧菌通过结合转导,得到含有重组质粒pds132-1的重组副溶血弧菌/pds132-1,并将重组副溶血弧菌/pds132-1接种至含有含庆大霉素的蔗糖培养基中进行培养;
12.(5)将含有sacb基因的上述敲除质粒potc导入大肠杆菌感受态,得到重组大肠杆菌/potc-sb;将重组大肠杆菌/potc-sb与步骤(4)中培养后的重组副溶血弧菌通过结合转导后,接种至含有氯霉素的培养基中进行培养,得到敲除了目的基因的副溶血弧菌。
13.在本发明的一种实施方式中,步骤(1)中,所述loxl-gm-loxr片段的核苷酸序列如seq id no.3所示。
14.在本发明的一种实施方式中,步骤(3)和(5)中,所述大肠杆菌包括但不限于cc118(λpir)、s17-1(λpir)。
15.在本发明的一种实施方式中,步骤(4)中,所述含有庆大霉素的蔗糖培养基为lb培养基,所述蔗糖的添加量至少为:10%(w/v)。
16.在本发明的一种实施方式中,所述庆大霉素的添加量为:10~30mg/l。
17.在本发明的一种实施方式中,所述含有氯霉素的培养基为lb培养基,所述氯霉素的添加量为:6~30mg/l。
18.在本发明的一种实施方式中,所述目的基因为副溶血弧菌基因组上任一不影响其生长的基因。
19.在本发明的一种实施方式中,所述目的基因包括但不限于ncbi上的gene bank登录号为vp_rs18950的蛋白、ncbi上的gene bank登录号为vp_rs17740的蛋白、ncbi上的gene bank登录号为vp_rs16510的蛋白、ncbi上的gene bank登录号为vp_rs06135的蛋白、ncbi上的gene bank登录号为vp_rs02520的蛋白、ncbi上的gene bank登录号为vp_rs11975的蛋白、ncbi上的gene bank登录号为vp_rs22195的蛋白、ncbi上的gene bank登录号为vp_rs23020的蛋白、ncbi上的gene bank登录号为vp_rs16800的蛋白、ncbi上的gene bank登录号为vp_rs16465的蛋白、ncbi上的gene bank登录号为vp_rs20840的蛋白、ncbi上的gene bank登录号为vp_rs03765的蛋白、ncbi上的gene bank登录号为vp_rs11205的转录因子、rpoe。
20.在本发明的一种实施方式中,所述rpoe基因在ncbi上的gene bank登录号为:vp_rs12550。rpoe表达的σe因子对细菌胞质外压力响应起到重要的作用(hews c l,cho t,rowley g,et al.maintaining integrity under stress:envelope stress response regulation of pathogenesis in gram-negative bacteria[j].front cell infect microbiol,2019,9:313.)。在高温下,rpoe是大肠杆菌中的必须基因(de las penas a,connolly l,gross c a.sigmae is an essential sigma factor in escherichia coli[j].j bacteriol,1997,179(21):6862-6864.),故敲除难度较大。
[0021]
在本发明的一种实施方式中,所述副溶血弧菌包括但不限于副溶血弧菌atcc33846。
[0022]
本发明还提供了上述敲除质粒在敲除副溶血弧菌基因组上任一不影响其生长的基因中的应用。
[0023]
有益效果
[0024]
(1)本发明提供了一种利用3种已有载体上的基因片段,经过重叠pcr与一步克隆
构建副溶血弧菌敲除工具质粒,并将该质粒用于构建副溶血弧菌基因敲除株及基因功能的研究。
[0025]
(2)传统副溶血弧菌敲除方法在第二次同源重组时具有理论上50%的敲除成功概率。本发明引入庆大抗性基因,在第二次同源重组时加入庆大霉素,通过抗性基因筛除第二次同源重组时回复为野生型的菌株,提高正确菌株的筛选效率,筛除回复突变的效率为100%。同时该方法可以减少挑取菌落的数量和时间,节约配置验证体系的时间,节省验证酶的用量,能够快速得到基因敲除突变菌株。
附图说明
[0026]
图1:副溶血弧菌引入potc质粒优化后的流程图。
[0027]
图2:敲除质粒potc与potc-sb的构建流程图。
[0028]
图3:potc与potc-sb的构建验证;其中,泳道1、2分别为potc的bstz17i、ecori酶切验证;泳道3、4分别为potc-sb的bstz17i、ecori酶切验证;m代表250-10000bp的dna marker。
[0029]
图4:使用优化体系敲除rpoe基因的第二次同源重组后的琼脂糖凝胶验证电泳图其中,m代表250-5000bp的dna marker,c代表菌落。
[0030]
图5:使用原始体系敲除rpoe基因的第二次同源重组后的琼脂糖凝胶验证电泳图;m代表250-10000bp的dna marker,g代表野生型基因组对照,p代表上下游同源臂对照,c代表菌落。
[0031]
图6:平板划线验证rpoe敲除后gm基因的去除。
[0032]
图7:平板划线验证rpoe敲除后质粒potc-sb的去除。
[0033]
图8:使用优化的副溶血弧菌敲除系统对外膜蛋白进行敲除,图为琼脂糖凝胶电泳验证敲除结果;其中,泳道1为野生型atcc33846中8个外膜蛋白基因,泳道2-9依次为δvp_rs11975、δvp_rs22195、δvp_rs23020、δvp_rs16800、δvp_rs16465、δvp_rs20840、δvp_rs03765、δvp_rs11205;m代表250-5000bp的dna marker。
具体实施方式
[0034]
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
[0035]
除非特别说明,以下实施例所用试剂和材料均为市售商品或者可以通过已知方法制备。
[0036]
本发明的方法适合于所有的副溶血弧菌,下述实施例仅以副溶血弧菌atcc33846为例进行说明,但其并不能对于本发明的保护范围进行限定。
[0037]
下述实施例中涉及的培养基如下:
[0038]
培养基均使用ddh2o配制,配制完成后121℃灭菌15~20min。
[0039]
lb培养基(g/l):酵母粉5,蛋白胨10,nacl 10。
[0040]
下述实施例中所涉及的引物序列如表1所示:
[0041]
表1下述实施例涉及的引物序列
[0042][0043][0044]
下述实施例中所涉及的菌株和质粒的来源如表2所示:
[0045]
表2下述实施例中涉及的菌株和质粒
[0046][0047]
实施例1:敲除质粒的构建
[0048]
具体步骤如下:
[0049]
1、质粒potc的构建
[0050]
(1)使用引物cm-p15a+与cm-p15a-将含有cmr与p15a基因的片段从pacyc184上扩增出来。使用pcr产物回收试剂盒回收pcr产物,用spei酶切消化。
[0051]
(2)使用引物traj-orit+与traj-orit-将traj与orit基因片段从pds132上扩增下来,使用pcr产物回收试剂盒纯化扩增得到的dna片段,用spei酶切消化。
[0052]
(3)使用引物ptac-cre+与ptac-cre-将cre基因片段从pdtw109上扩增下来,使用pcr产物回收试剂盒回收pcr产物。
[0053]
(4)对步骤(1)和(2)中的酶切产物各取1μg配置连接体系,进行4℃过夜连接,70℃处理10min使连接酶失活。
[0054]
(5)将步骤(3)中的dna片段与步骤(4)中的连接产物使用一步克隆试剂盒进行连接,转化至cc118(λpir)感受态在37℃条件下培养24h,挑取菌落验证后提取质粒测序(结果如图3所示),测序正确即为本发明的敲除质粒,将该质粒命名为potc。
[0055]
选取测序结果正确的质粒potc电转化至s17-1(λpir),菌落验证正确后在lb液体培养基中,在37℃条件下进行培养12h后,取800μl菌液加入甘油管保种于-70℃。
[0056]
2、质粒potc-sb的构建
[0057]
(1)使用引物sacb+和sacb-将sacb片段从pds132上扩增,得到sacb片段;使用bstz17i对potc质粒酶切线性化,最后使用一步克隆试剂盒将sacb片段与线性化质粒potc片段连接,并化转入cc118(λpir),进行菌落pcr验证并测序(结果如图3所示)。
[0058]
(2)将测序正确的质粒potc-sb电转入s17-1(λpir),菌落验证正确后在lb液体培养基中37℃条件下进行培养12h后,取800μl菌液加入甘油管保种于-70℃。
[0059]
potc-sb质粒构建流程如图2所示,构建完成后使用快切酶bstz17i、ecori进行酶切验证,结果显示:含有sacb基因的potc-sb重组质粒构建成功,同时得到含有质粒potc-sb的s17-1(λpir)。
[0060]
实施例2:副溶血弧菌基因缺失突变株的构建
[0061]
副溶血弧菌自身具有同源重组系统。利用这一点,含有同源臂的自杀质粒进入细胞后会整合到基因组上,本技术以其序列的ncbi上登录号依次为vp_rs11975、vp_rs22195、vp_rs23020、vp_rs16800、vp_rs16465、vp_rs20840、vp_rs03765、vp_rs11205的基因,以及ncbi上登录号为vp_rs12550的rpoe为例,证明本发明的敲除方法可高效成功的实施。
[0062]
具体步骤如下:
[0063]
1、基因敲除菌株的构建
[0064]
(1)利用表1中vp_rs11975、vp_rs22195、vp_rs23020、vp_rs16800、vp_rs16465、vp_rs20840、vp_rs03765、vp_rs11205、rpoe的上下游同源臂引物进行pcr扩增,分别得到各个基因的上游和下游同源臂。
[0065]
(2)使用引物gm-和gm+从pwjw101扩增loxl-gm-loxr片段。
[0066]
(3)通过融合pcr,分别将步骤(1)和步骤(2)的各个基因的上游同源臂、loxl-gm-loxr、各个基因的下游同源臂整合到一起,分别得到含有各个基因的同源臂的up-loxl-gm-loxr-down。
[0067]
(4)分别将pds132质粒与步骤(3)得到的含有各个基因的同源臂的up-loxl-gm-loxr-down使用xbai或sali酶切(其中,vp_rs23020同源臂使用一步克隆试剂盒与酶切线性化的pds132质粒连接),分别得到不同的连接产物;
[0068]
分别将上述连接产物在4℃过夜连接后转化进入cc118(λpir),涂布于含有浓度为30mg/l庆大霉素的lb平板;通过菌落pcr验证,挑取正确的菌37℃条件下培养24h后,分别提取质粒。通过电转化分别将上述质粒导入大肠杆菌s17-1(λpir),涂布于含有浓度为30mg/l庆大霉素的lb平板。
[0069]
(5)第一次接合转导:
[0070]
分别将步骤(4)得到的含有庆大霉素的lb平板的大肠杆菌s17-1(λpir)与副溶血弧菌atcc33846分别在37℃条件下培养至od
600
约为1.0,各取1ml菌液用lb液体培养基清洗两次后,100μl lb液体培养基共悬浮,点种于lb平板上,正置过夜培养。分别使用灭菌棉签刮下菌苔悬于1ml lb中,涂布于含有浓度为10mg/l多粘菌素b与含有浓度为6mg/l氯霉素的lb平板。
[0071]
在第一次接合转导时发生第一次同源重组,分别挑取上述lb平板上的单菌落进行菌落pcr并培养提取基因组验证。验证正确的菌落应具有两条条带,一条长度为质粒上同源臂和庆大抗性基因的长度,另一条长度为基因组上同源臂与目标基因的长度。
[0072]
(6)蔗糖筛选:
[0073]
分别将步骤(5)验证正确的菌接种于含浓度为30mg/l庆大霉素及含有10%(w/v)蔗糖的lb培养基的试管中,在37℃条件下进行培养12h后,分别取50μl涂布于含浓度为30mg/l庆大霉素及含有10%(w/v)蔗糖的lb平板上,对单菌落进行菌落pcr和基因组验证,此时,在各个不同的缺失突变构建株中分别发生了第二次同源重组,pds132质粒带着目标基因从副溶血弧菌atcc33846基因组上去掉,分别得到含有庆大霉素抗性的缺失了相应蛋白的基因缺失突变菌株。
[0074]
(7)第二次接合转导:
[0075]
分别将实施例1的步骤2得到的含有质粒potc-sb的s17-1(λpir)与本方法第(6)步得到的基因缺失突变株,按照上述第(5)步的接合转导试验方法进行接合,分别得到含有质粒potc-sb的基因缺失突变株。
[0076]
分别挑取上述单菌落进行验证,此时会有三种结果:
[0077]
第一种为只有一条长度为上下游同源臂和庆大抗性基因的条带;
[0078]
第二种为只有一条长度为比上下游同源臂大100bp的条带,即已去除庆大抗性基因;
[0079]
第三种为出现以上两种条带,即菌落未完全去除pds132质粒。
[0080]
(8)对于第一种和第三种的菌落,挑取并置于含有浓度为30mg/l的氯霉素抗性的lb液体中37℃条件下培养12h,使cre酶进行更彻底的切除,培养结束后,取50-200μl菌液再次涂布于含有浓度为30mg/l的氯霉素平板进行单菌落验证。
[0081]
对于第二种菌落,划线于含有浓度为10mg/l的庆大霉素lb平板进行负筛选。将未在该庆大霉素平板上长出的菌落,接于含有10%(w/v)蔗糖lb液体培养基试管中过夜去除potc-sb质粒,取50μl上述去除potc-sb质粒的菌液,涂布于含有10%(w/v)蔗糖lb平板上,挑取单菌落划线于含有浓度为30mg/l的氯霉素lb平板进行负筛选。未长出的菌落为正确的敲除完成菌株。
[0082]
2、结果的表述:
[0083]
(1)优化的敲除系统中,在上下游同源臂之间引入庆大抗性基因。为了敲除完成后顺利去除庆大抗性基因,引入cre/loxp系统,loxp位点位于庆大抗性基因两端(图1)。cre酶能够识别并剪去loxp位点。为了使cre基因进入副溶血弧菌atcc33846细胞中,构建了potc-sb质粒(图2),质粒构建完成后进行验证(图3)。
[0084]
(2)本发明选取rpoe基因验证优化的敲除体系。在优化的敲除体系的第二次同源重组后,成功敲除的条带长度应为上下游同源臂与庆大基因的总和,为2268bp。未成功的条带长度为上下游同源臂与靶基因的总和,为1756bp。
[0085]
由图4可知,随机挑取的10个单菌落中,敲除成功1个,质粒未完全去除9个。
[0086]
而在原始敲除体系的第二次同源重组后,成功敲除的条带长度应为上下游同源臂,为1178bp。未成功的条带长度为上下游同源臂与靶基因的总和,为1756bp。由图5可知,随机挑取的10个单菌落全部回复为野生型。优化的敲除体系可以筛除回复为野生型的菌体。
[0087]
(3)本发明在靶基因敲除后,进行庆大抗性基因的去除。由图6可知,导入质粒potc-sb后,cre酶对loxp位点间的庆大抗性基因的去除效率,25个单菌落平板划线后有5株去除庆大抗性基因,效率为20%。
[0088]
而由图7可知,10%蔗糖环境去除potc-sb质粒的效率,25个单菌落平板划线后有25株均已去除potc-sb质粒,效率为100%。
[0089]
(4)使用优化的敲除体系一次性对8个基因进行敲除。最终的敲除株均成功构建(图8)。说明优化的敲除体系较原始具有更高的结果稳定性,减少重复多遍的第二次同源重组。
[0090]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1