一种构建碳-氮轴手性吲哚-萘酚联芳基化合物的方法
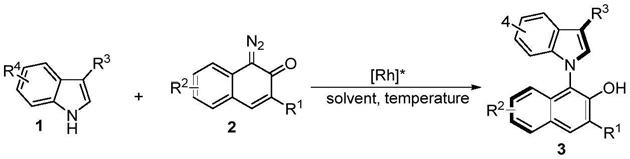
1.本发明属于不对称催化领域,具体的说是一种手性铑催化吲哚衍生物的n-h键插入1-重氮-2-萘酮铑卡宾构建碳-氮轴手性吲哚-萘酚联芳基化合物的方法。
背景技术:
2.轴手性配体和催化剂十分普遍,其在不对称催化中有着重要的应用,如binap已实现多个不对称催化反应的工业化应用,轴手性磷酸在有机小分子催化中展现了出色的催化性能和应用前景。构建轴手性骨架的研究正受到越来越多的关注。
3.除了碳-碳轴手性化合物,碳-杂原子轴手性也广泛存在,比如marinopryyole,murrastifoline-f,eupolyphagin结构中均含有碳-氮手性轴。然而,相对于碳-碳轴手性的研究,碳-杂原子轴手性的合成方法却很少,已报道的方法有:取代苯胺氮的官能化(zhang,l.;zhang,j.;ma,j.;cheng,d.-j.;tan,b.j.am.chem.soc.2017,139,1714.);n-芳基杂环化合物的芳基官能化(zhang,j.;xu,q.;wu,j.;fan,j.;xie,m.org.lett.2019,21,6361.);手性磷酸催化的萘基偶氮羧酸酯和咔唑的不对称胺化(xia,w.;an,q.-j.;xiang,s.-h.;li,s.;wang,y.-b.;tan,t.angew.chem.int.ed.2020,59,6775.)。
4.卡宾的n-h键插入反应一种高效构建c-n键的方法,如果能通过合适的手性催化剂催化卡宾插入芳杂环的n-h键来构建c-n轴手性,将是一种非常直接有效的方法。本发明提供了一种手性铑催化吲哚衍生物n-h键插入1-重氮-2-萘酮合成碳-氮轴手性吲哚-萘酚联芳基化合物的新方法。
技术实现要素:
5.本发明公开了一种手性铑催化吲哚衍生物n-h键插入1-重氮-2-萘酮合成碳-氮轴手性吲哚-萘酚联芳基化合物的方法,该方法以优秀的产率和对应选择性得到吲哚-萘酚类碳-氮轴手性联芳基化合物。反应经历的可能过程:1-重氮-2-萘酮在铑催化下生成手性铑卡宾,受到吲哚氮原子进攻生成叶立德中间体,随后经过立体选择性的质子迁移、芳构化构筑碳-氮手性轴。
6.本发明具体的反应通式如下:
[0007][0008]
一种构建碳-氮轴手性吲哚-萘酚联芳基化合物的方法,具体按照下述步骤进行:氩气保护下,向反应管中加入手性铑催化剂、吲哚衍生物(1)、1-重氮-2-萘酮(2)及溶剂,之后搅拌反应一定时间,得到吲哚-萘酚碳-氮轴手性联芳基化合物(3)。
[0009]
对本发明内容的具体说明如下:
[0010]
其中手性铑催化剂为:rh2(s-pta)4,rh2(s-ptv)4,rh2(s-pttl)4,rh2(s-tfpttl)4,
rh2(s-tcpttl)4,rh2(s-tbpttl)4,rh2(s-tcptad)4,rh2(s-ptad)4,rh2(s-nttl)4,rh2(s-bp)4,rh2(s-bsp)4,最优的催化剂为:rh2(s-nttl)4。
[0011]
手性铑催化剂结构为:
[0012][0013]
碳-氮轴手性吲哚-萘酚联芳基化合物(3)结构可以为:
[0014][0015]
a1=氢;a2=乙基、异丙基、叔丁基、叔戊基、(2-甲基-丁-3-烯-2-基)、2-苯基-丙-2-基或(2-(萘-2-基)-丙-2-基);
[0016]
a3=甲基、乙基、苯基或氢;a4=甲基、乙基、叔丁基、苯基、卤素或氢;
[0017]
a5=甲基、乙基、异丙基、叔丁基或卤素;a6=甲基或氢;
[0018]
碳-氮轴手性联芳基化合物(3)结构中吲萘环上的取代基为:
[0019][0020]
b1=氢、甲基、烷氧羰基、卤素、甲氧基或乙氧基;b2=氢或溴;b3=氢、溴或烷氧羰基;b4=氢、溴、甲基、苯基或烷氧羰基;b5=氢、溴、甲基或苯基;b6=氢。
[0021]
构建的氮轴手性吲哚-萘酚联芳基化合物(3)结构有:
[0022]
[0023][0024]
反应的溶剂为:二氯甲烷,二氯乙烷,氯仿,甲苯,甲基叔丁基醚等,其中最优的溶剂为二氯甲烷。
[0025]
反应中手性铑催化剂、吲哚(1)、1-重氮-2-萘酮(2)的摩尔比为:0.01~0.02:1:1.1~2.5,最优的摩尔比为:0.01:1:1.3。
[0026]
反应液浓度为:吲哚衍生物(1)浓度为0.025~0.2mol/l,最优的反应浓度:0.1mol/l。
[0027]
反应的温度为:0-40℃,其中最佳温度为30℃。
[0028]
反应的时间为:2-8小时。进一步优选的反应时间为4h。
[0029]
碳-氮轴手性吲哚-萘酚联芳基化合物经过衍生合成了含有c-n轴手性的单膦配体(如l1),用于钯催化不对称烯丙基化反应中。
[0030][0031]
有益效果
[0032]
本发明公开了一种手性铑催化吲哚衍生物的n-h键插入金属卡宾构建碳-氮轴手性联芳基化合物的方法,通过卡宾转移策略一步高效完成轴手性的构建,本发明方法的优点有:反应条件温和、操作简单、底物普适性好、反应产率高、对映选择性好。
[0033]
本发明中的碳-氮轴手性化合物可以作为开发新型催化剂、配体的前体,其结构中取代基位置和电性的兼容性也有利于对衍生轴手性配体的结构进行修饰。本发明通过简单衍生得到了c-n轴手性单膦配体(l1),在钯催化不对称烯丙基化反应中具有一定的催化活
性。
附图说明
[0034]
图1为实施例1得到的3aa的1h-nmr(核磁氢谱);
[0035]
图2为实施例1得到的3aa的
13
c-nmr(核磁碳谱);
[0036]
图3为实施例1得到的3aa的hrms(高分辨质谱);
[0037]
图4为实施例1得到的3aa的hplc(高效液相色谱)。
具体实施方式
[0038]
下面将通过具体实施例对本发明做进一步说明,本发明并不局限于以下的实施例:
[0039]
实施例1:
[0040][0041]
氩气保护下,向反应管中加入rh2(s-nttl)4(2.9mg,0.002mmol),1a(39.8mg,0.2mmol),2a(59.3mg,0.26mmol)和dcm(2ml),反应在30℃搅拌4小时。反应液减压蒸除溶剂、柱层析纯化(淋洗剂为石油醚:乙酸乙酯=100:1-50:1)得到黄色固体产物3aa(69.6mg,收率87%,92%ee),熔点:74-76℃。3aa结构表征见表1。
[0042]
实施例2:
[0043][0044]
氩气保护下,向反应管中加入rh2(s-pttl)4(2.5mg,0.002mmol),1a(39.8mg,0.2mmol),2a(68.4mg,0.3mmol)和dcm(2ml),反应在30℃搅拌4小时。反应液减压蒸除溶剂、柱层析纯化(淋洗剂为石油醚:乙酸乙酯=100:1-50:1)得到黄色固体产物3aa(56.7mg,收率71%,79%ee)。
[0045]
实施例3:
[0046][0047]
氩气保护下,向反应管中加入rh2(s-nttl)4(2.9mg,0.002mmol),1a(39.8mg,0.2mmol),2a(68.4mg,0.3mmol)和dce(2ml),反应在30℃搅拌4小时。反应液减压蒸除溶剂、
柱层析纯化(淋洗剂为石油醚:乙酸乙酯=100:1-50:1)得到黄色固体产物3aa(53.5mg,收率67%,91%ee)。
[0048]
实施例4:
[0049][0050]
氩气保护下,向反应管中加入rh2(s-nttl)4(2.9mg,0.002mmol),1a(39.8mg,0.2mmol),2a(68.4mg,0.3mmol)和dcm(2ml),反应在0℃搅拌4小时。反应液减压蒸除溶剂、柱层析纯化(淋洗剂为石油醚:乙酸乙酯=100:1-50:1)得到黄色固体产物3aa(28.8mg,收率36%,93%ee)。
[0051]
实施例5:
[0052][0053]
氩气保护下,向反应管中加入rh2(s-nttl)4(2.9mg,0.002mmol),1a(39.8mg,0.2mmol),2a(59.3mg,0.26mmol)和dcm(8ml),反应在30℃搅拌4小时。反应液减压蒸除溶剂、柱层析纯化(淋洗剂为石油醚:乙酸乙酯=100:1-50:1)得到黄色固体产物3aa(62.3mg,收率78%,92%ee)。
[0054]
实施例6:
[0055][0056]
将1a(39.8mg,0.2mmol)与2b(44.2mg,0.26mmol)通过实施例1的方法反应1小时,得到褐色油状产物3ab(50.0mg,收率73%,86%ee)。3ab结构表征见表1。
[0057]
实施例7:
[0058][0059]
将1a(39.8mg,0.2mmol)与2c(62.9mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ac(73.7mg,收率89%,88%ee),熔点:73-75℃。3ac结构表征见表1。
[0060]
实施例8:
[0061][0062]
将1a(39.8mg,0.2mmol)与2d(79.0mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ad(60.0mg,收率63%,73%ee),熔点:141-143℃。3ad结构表征见表1。
[0063]
实施例9:
[0064][0065]
将1a(39.8mg,0.2mmol)与2e(53.0mg,0.26mmol)通过实施例1的方法,得到褐色油状产物3ae(61.3mg,收率86%,84%ee)。3ae结构表征见表1。
[0066]
实施例10:
[0067][0068]
将1a(39.8mg,0.2mmol)与2f(47.8mg,0.26mmol)通过实施例1的方法,得到褐色油状产物3af(57.2mg,收率76%,92%ee)。3af结构表征见表1。
[0069]
实施例11:
[0070][0071]
将1a(39.8mg,0.2mmol)与2g(73.0mg,0.26mmol)通过实施例1的方法,得到褐色油状产物3ag(52.0mg,收率65%,87%ee)。3ag结构表征见表1。
[0072]
实施例12:
[0073]
[0074]
将1a(39.8mg,0.2mmol)与2h(47.8mg,0.26mmol)通过实施例1的方法,得到褐色油状产物3ah(47.0mg,收率66%,93%ee)。3ah结构表征见表1。
[0075]
实施例13:
[0076][0077]
将1a(39.8mg,0.2mmol)与2i(89.2mg,0.26mmol)通过实施例1的方法,得到白色固体产物3ai(58.8mg,收率70%,86%ee),熔点:151-153℃。3ai结构表征见表1。
[0078]
实施例14:
[0079][0080]
将1a(39.8mg,0.2mmol)与2j(62.9mg,0.26mmol)通过实施例1的方法,得到黄色油状产物3aj(58.0mg,收率70%,92%ee)。3aj结构表征见表1。
[0081]
实施例15:
[0082][0083]
将1a(39.8mg,0.2mmol)与2k(79.6mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ak(70.8mg,收率74%,93%ee),熔点:102-104℃。3ak结构表征见表1。
[0084]
实施例16:
[0085][0086]
将1a(39.8mg,0.2mmol)与2l(79.1mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3al(69.5mg,收率73%,92%ee),熔点:163-165℃。3al结构表征见表1。
[0087]
实施例17:
[0088][0089]
将1b(42.6mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ba(63.0mg,收率76%,91%ee),熔点:124-125℃。3ba结构表征见表1。
[0090]
实施例18:
[0091][0092]
将1c(45.4mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ca(70.2mg,收率82%,85%ee),熔点:155-157℃。3ca结构表征见表1。
[0093]
实施例19:
[0094][0095]
将1d(48.2mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3da(68.1mg,收率77%,91%ee),熔点:183-185℃。3da结构表征见表1。
[0096]
实施例20:
[0097][0098]
将1e(52.2mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到白色固体产物3ea(68.4mg,收率74%,94%ee),熔点:223-224℃。3ea结构表征见表1。
[0099]
实施例21:
[0100][0101]
将1f(43.0mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固
体产物3fa(69.1mg,收率83%,96%ee),熔点:130-132℃。3fa结构表征见表1。
[0102]
实施例22:
[0103][0104]
将1g(42.6mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ga(67.1mg,收率81%,90%ee),熔点:66-68℃。3ga结构表征见表1。
[0105]
实施例23:
[0106][0107]
将1h(40.2mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ha(71.6mg,收率89%,90%ee),熔点:161-163℃。3ha结构表征见表1。
[0108]
实施例24:
[0109][0110]
将1i(37.4mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色油状产物3ia(69.1mg,收率89%,91%ee)。3ia结构表征见表1。
[0111]
实施例25:
[0112][0113]
将1j(50.2mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色油状产物3ja(79.6mg,收率88%,92%ee)。3ja结构表征见表1。
[0114]
实施例26:
[0115][0116]
将1k(37.4mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3ka(38.8mg,收率65%,93%ee),熔点:163-165℃。3ka结构表征见表1。
[0117]
实施例27:
[0118][0119]
将1l(49.8mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色固体产物3la(77.4mg,收率86%,90%ee),熔点:145-147℃。3la结构表征见表1。
[0120]
实施例28:
[0121][0122]
将1m(62.6mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色油状产物3ma(85.3mg,收率83%,90%ee)。3ma结构表征见表1。
[0123]
实施例29:
[0124][0125]
将1n(59.8mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,得到黄色油状产物3na(90.0mg,收率90%,89%ee)。3na结构表征见表1。
[0126]
实施案例30:
[0127][0128]
将1o(26.2mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,未得到目标
产物。
[0129]
实施案例31:
[0130][0131]
将1p(53.9mg,0.2mmol)与2a(59.3mg,0.26mmol)通过实施例1的方法,未得到目标产物。
[0132]
实施例32:
[0133][0134]
将1a(39.8mg,0.2mmol)与2b(44.2mg,0.26mmol)通过实施例1的方法反应8小时,得到褐色油状产物3ab(50.0mg,收率72%,80%ee)。3ab结构表征见表1。
[0135]
案例说明,想要获得目标c-n轴手性化合物,吲哚化合物在手性轴的两侧必须同时存在取代基,以提供位阻。否则无法得到碳-氮轴手性吲哚-萘酚联芳基化合物。且反应时间的延长会导致ee值有所降低。
[0136]
实施例33:c-n轴手性膦配体(l1)的合成
[0137][0138]
(第一步):氩气保护下,向反应管中加入rh2(s-nttl)4(5.8mg,0.004mmol),1a(79.6mg,0.4mmol),2b(88.4mg,0.52mmol)和dcm(2ml),反应在30℃搅拌4小时后,向反应中dmap(13mg,0.1mmol)和吡啶(123mg,1.56mmol)和dcm(5.0ml),随后在0℃缓慢加入tf2o(440.0mg,1.56mmol),反应混合液升到室温搅拌反应2小时。反应混合物用水(10ml)淬灭、二氯甲烷(15ml)萃取两次;合并的有机相用食盐水洗涤、干燥减压蒸掉溶剂、残余物通过柱层析纯化(石油醚:乙酸乙酯=100:1~60:1)得到白色固体产物4(136.6mg,收率72%),熔点:106-108℃。1h nmr(300mhz,cdcl3)δ7.94(d,j=9.1hz,1h),7.89(d,j=8.2hz,1h),7.60(d,j=8.2hz,1h),7.54-7.45(m,2h),7.42-7.35(m,1h),7.30(d,j=8.5hz,1h),6.94-6.85(m,2h),6.50(s,1h),6.11(dd,j=17.4,10.5hz,1h),5.06(dd,j=17.4,1.3hz,1h),4.99(dd,j=10.5,1.3hz,1h),2.22(s,3h),1.58-1.43(m,6h).
13
cnmr(75mhz,cdcl3)δ147.5,143.5,139.2,133.2,132.2,132.0,130.4,128.7,128.5,128.3,127.8,122.6(q,j=318.5hz),125.6,124.9,124.8,124.4,121.5,121.4,119.8,111.2,110.7,37.7,28.2,28.1,21.7.
19
f nmr(282mhz,cdcl3)δ-74.0(3f).hrms(esi):calculated for c
25h23
f3no3s[m+h]
+
:474.1345;found:474.1350.
[0139]
(第二步):氩气氛围下,向干燥的反应管中加入4(94.7mg,0.2mmol)、dmso(6ml)、ph2p(o)h(161.6mg,0.8mmol)、pd(oac)2(4.5mg,0.02mmol)和dppb(10.2mg,0.024mmol),充分超声去氧后,再加入net3(122.3mg,1.2mmol),之后反应在80℃油浴中搅拌2小时。反应混合物冷却后用1m稀盐酸(10ml)淬灭、二氯甲烷(15ml)萃取两次;合并的有机相依次用饱和碳酸氢钠(15ml)洗涤和食盐水(15ml)洗涤、干燥后减压蒸掉溶剂、残余物通过柱层析纯化(石油醚:乙酸乙酯=20:1~2:1)得到淡黄色油状产物5(96.8mg,收率92%,91%ee)。hplc检测条件:daicel chiralpak ia column,n-hexane/i-proh=85/15,flow rate 1ml/min,λ=225nm,tr=7.72min(major)and 9.71min(minor).[α]
d20
:+50.0(c=1.00,chcl3;91%ee).1h nmr(400mhz,cdcl3)δ8.06-7.93(m,3h),7.59-7.46(m,5h),7.38-7.27(m,3h),7.23-7.10(m,5h),6.86(s,1h),6.75-6.65(m,2h),6.07(dd,j=17.4,10.5hz,1h),5.92(s,1h),5.07(d,j=17.4hz,1h),5.01(d,j=10.5hz,1h),2.09(s,3h),1.44-1.37(m,6h).
13
c nmr(100mhz,cdcl3)δ147.7,140.5,140.4,139.7,136.0,132.2,132.1,131.9,131.8,131.7,131.4,131.19,131.16,131.1,131.0,130.92,130.89,130.8,130.1,128.9,128.8,128.7,128.5,128.11,128.06,127.9,127.8,127.7,126.9,124.5,123.71,123.65,120.8,120.5,111.5,110.5,37.4,28.1,27.7,21.4.
31
p nmr(121mhz,cdcl3)δ25.93.hrms(esi):calculated for c
36h33
nop[m+h]
+
:526.2294;found:526.2286.
[0140]
(第三步):氩气氛围下,在0℃向干燥的反应管中依次加入5(52.6mg,0.1mmol,91%ee)、甲苯(2ml)、et3n(71.4mg,0.7mmol)和hsicl3(67.2mg,0.5mmol),之后反应在80℃加热模块上搅拌2.5小时。反应混合物冷却后用乙醚(15ml)稀释,加入碳酸钠溶液(15ml)淬灭,经一层硅藻土过滤,母液分离出有机相、干燥、浓缩,残余物通过柱层析纯化(石油醚:乙酸乙酯=100:1~20:1)得到白色固体产物l1(46.4mg,收率91%,92%ee),熔点:152-154℃。hplc检测条件:daicel chiralpak ia column,n-hexane/i-proh=99.8/0.2,flow rate 0.6ml/min,λ=225nm,tr=26.89min(minor)and 32.36min(major).[α]
d20
:+137.0(c=1.00,chcl3;92%ee).1h nmr(400mhz,cdcl3)δ7.92-7.82(m,2h),7.62(d,j=8.1hz,1h),7.52-7.45(m,1h),7.35-7.15(m,12h),7.04(d,j=8.4hz,1h),6.91(d,j=8.1hz,1h),6.49(s,1h),6.43(s,1h),6.08(dd,j=17.4,10.5hz,1h),5.07(d,j=17.4hz,1h),5.00(d,j=10.5hz,1h),2.25(s,3h),1.41-1.31(m,6h).
13
c nmr(100mhz,cdcl3)δ147.9,140.8,140.5,139.4,137.6,137.5,136.9,136.78,136.76,136.6,134.41,134.35,134.1,134.0,133.8,132.09,132.05,131.4,129.6,129.0,128.64,128.62,128.57,128.5,128.4,128.0,127.5,127.3,126.41,126.39,124.1,123.93,123.90,123.4,121.03,120.95,111.1,110.8,37.5,28.1,28.0,21.7.
31
p nmr(121mhz,cdcl3)δ-15.82.hrms(esi):calculated for c
36h33
np[m+h]
+
:510.2345;found:510.2346.
[0141]
实施例34:c-n轴手性膦配体(l1)催化应用
[0142][0143]
氩气氛围下,向l1(11.8mg,0.02mmol)、[pd(η
3-c3h5)cl]2(2.9mg,0.01mmol)、lioac(1.1mg,0.016mmol)的二氯甲烷(1ml)混合物中加入n,o-双三甲硅基乙酰胺(bsa,122.0mg,0.6mmol)and 6(50.4mg,0.2mmol),搅拌30分钟后加入丙二酸二乙酯(96.0mg,
0.6mmol)。反应混合物室温搅拌反应24小时。反应混合物用乙醚(15ml)和水(15ml)稀释,分出有机相、干燥、浓缩,残余物通过柱层析纯化(石油醚:乙酸乙酯=50:1~20:1)得到产物8(21.8mg,收率31%,27%ee)。hplc检测条件:daicel chiralpak iacolumn,n-hexane/i-proh=85/15,flow rate 1ml/min,λ=225nm,tr=8.08min(minor)and 10.56min(major).[α]
d20
:-6.3(c=1.0,chcl3;27%ee).1hnmr(400mhz,cdcl3)δ7.43-7.11(m,10h),6.48(d,j=16.0hz,1h),6.34(dd,j=16.0,8.8hz,1h),4.27(dd,j=11.2,8.8hz,1h),4.16(q,j=7.2hz,2h),4.06-3.84(m,3h),1.20(t,j=7.2hz,3h),1.00(t,j=7.2hz,3h).
13
cnmr(100mhz,cdcl3)δ167.9,167.5,140.4,136.9,131.7,129.4,128.7,128.5,128.0,127.6,127.2,126.4,61.6,61.4,57.8,49.3,14.2,13.8.
[0144]
能合理预测到其它化合物的也能合成c-n轴手性单膦配体,在钯催化不对称烯丙基化反应中具有一定的催化活性,具有潜在应用价值。
[0145]
实施例35:吲哚化合物1c~1e,1g的制备
[0146][0147]
在氩气保护下,向一个干燥的烧瓶中加入相应取代基的吲哚(1mmol,1.0equiv),pd(pph3)4(0.01mmol,0.01equiv),4ml的干燥四氢呋喃和2-甲基-3-丁烯-2-醇(10mmol,10equiv),之后缓慢加入bet3(2.4mmol,1m inhexane)。将反应液水浴升温至40℃反应7天。当相应取代基的吲哚消耗完,向反应液中加入适量乙酸乙酯,然后用饱和碳酸氢钠溶液洗涤有机相,收集有机相用无水硫酸钠干燥,然后减压蒸除溶剂、柱层析纯化,得到相应的吲哚化合物1c~1e,1g。
[0148][0149]
通过实施案例35得到无色油状吲哚1c(收率80%)。1h nmr(300mhz,cdcl3)δ7.73-7.56(m,2h),7.13(s,1h),7.02-6.94(m,1h),6.82(d,j=2.3hz,1h),6.21-6.03(m,1h),5.14-4.96(m,2h),3.06-2.92(m,1h),1.49(s,6h),1.32-1.27(m,6h).
13
c nmr(75mhz,cdcl3)δ147.9,142.8,137.5,124.3,123.8,121.1,119.7,118.4,110.6,108.5,37.6,34.3,28.1,24.6.hrms(esi):calculated for c
16h22
n[m+h]
+
:228.1747;found:228.1746.
[0150][0151]
通过实施案例35得到无色油状吲哚1d(收率78%)。1h nmr(300mhz,cdcl3)δ7.71-7.60(m,2h),7.29(d,j=1.4hz,1h),7.18-7.12(m,1h),6.83(d,j=2.4hz,1h),6.12(dd,j=17.4,10.5hz,1h),5.09(dd,j=17.4,1.3hz,1h),5.01(dd,j=10.5,1.3hz,1h),1.49(s,6h),1.37(s,9h).
13
c nmr(75mhz,cdcl3)δ148.0,145.1,137.4,123.8,123.7,120.8,119.9,
117.3,110.5,107.6,37.6,34.8,31.9,28.1.hrms(esi):calculated for c
17h22
n[m+h]
+
:242.1903;found:242.1899.
[0152][0153]
通过实施案例35得到白色固体吲哚1e(收率81%),熔点:73-75℃。1h nmr(400mhz,cdcl3)δ7.75(d,j=8.2hz,1h),7.65(s,1h),7.61-7.56(m,2h),7.43-7.36(m,3h),7.34-7.25(m,2h),6.86(s,1h),6.21-6.03(m,1h),5.14-4.97(m,2h),1.50(s,6h).
13
c nmr(100mhz,cdcl3)δ147.8,142.3,137.7,135.2,128.9,127.5,126.7,125.4,123.9,121.7,121.0,118.9,110.9,109.9,37.6,28.2.hrms(esi):calculated for c
19h20
n[m+h]
+
:262.1590;found:262.1583.
[0154][0155]
通过实施案例35得到无色油状吲哚1g(收率83%)。1h nmr(400mhz,cdcl3)δ7.51(s,1h),7.46(s,1h),7.03(s,1h),6.76(d,j=2.4hz,1h),6.23-6.00(m,1h),5.11-4.95(m,2h),2.33(s,6h),1.52-1.45(m,6h).
13
cnmr(100mhz,cdcl3)δ148.0,136.2,130.7,127.5,124.4,123.3,121.5,119.5,111.8,110.7,37.6,28.2,20.5.hrms(esi):calculated for c
15h20
n[m+h]
+
:214.1590;found:214.1587.
[0156]
实施例36:吲哚化合物1i的制备
[0157][0158]
在氩气保护情况下,向烧瓶中加入6-甲基吲哚(6mmol),四丁基碘化铵(3mmol),三氟甲磺酸锌(3.6mmol),二异丙基乙胺(6.6mmol)和18ml干燥的甲苯。将上述反应液在室温下搅拌15分钟后,缓慢滴加溴代叔丁烷(3mmol)。反应3小时后,通过薄层色谱点板分析监测反应。反应结束后,用饱和氯化铵水溶液淬灭反应,用乙酸乙酯和水萃取,收集有机相用无水硫酸钠干燥。旋蒸除去溶剂后经过柱层析法分离纯化得到无色油状吲哚1i(收率42%)。1h nmr(300mhz,cdcl3)δ7.69(d,j=8.2hz,1h),7.53(s,1h),7.05(s,1h),6.92(d,j=8.2hz,1h),6.76(d,j=2.4hz,1h),2.44(s,3h),1.43(s,9h).
13
c nmr(100mhz,cdcl3)δ137.7,131.3,126.7,123.8,121.0,120.6,118.7,111.4,31.7,30.9,21.7.hrms(esi):calculated for c
13h18
n[m+h]
+
:188.1434;found:188.1429.
[0159]
实施例37:吲哚化合物1k的制备
[0160][0161]
在氩气保护情况下,向干燥的烧瓶中加入3,5-二甲基苯肼盐酸盐(3mmol)和acoh(5ml),将反应液加热至70℃,用注射器将异戊醛(3mmol)加入到反应体系中。反应12小时后,通过薄层色谱点板监测反应。待原料消耗完全后,用乙酸乙酯和水萃取,收集有机相用无水硫酸钠干燥。旋蒸除去溶剂后经过柱层析法分离纯化得到黄色油状吲哚1k(收率32%)。1hnmr(400mhz,cdcl3)δ7.66(s,1h),6.92(s,1h),6.86(s,1h),6.68(s,1h),3.52-3.36(m,1h),2.67(s,3h),2.39(s,3h),1.35-1.28(m,6h).
13
c nmr(100mhz,cdcl3)δ137.3,131.6,130.4,125.4,123.2,123.1,118.6,109.0,26.0,25.0,21.5,20.4.hrms(esi):calculated for c
13h18
n[m+h]
+
:188.1434;found:188.1431.
[0162]
实施例38:吲哚化合物1l~1n的制备
[0163][0164]
向一只50ml烧瓶中加入pdcl2(mecn)2(0.25mmol),20ml的dce,以及相应的吲哚(5mmol)和相应的芳基醇(6mmol),之后将反应液升温至90℃反应12小时。12小时后,通过薄层色谱点板监测反应。待原料消耗完全后,用乙酸乙酯和水萃取,收集有机相用无水硫酸钠干燥。旋蒸除去溶剂后经过柱层析法分离纯化得到相应的吲哚1l~1n。
[0165][0166]
通过实施例38得到无色油状吲哚1l(收率72%)。1h nmr(400mhz,cdcl3)δ7.57(s,1h),7.37-7.29(m,2h),7.26-7.18(m,2h),7.17-7.10(m,1h),7.04(s,1h),6.96-6.87(m,2h),6.70(d,j=8.2hz,1h),2.37(s,3h),1.73(s,6h).
13
c nmr(100mhz,cdcl3)δ150.1,137.7,131.5,128.2,126.5,126.0,125.7,124.0,121.0,121.0,120.1,111.2,39.0,30.8,21.8.hrms(esi):calculated for c
18h20
n[m+h]
+
:250.1590;found:250.1584.
[0167][0168]
通过实施例38得到无色油状吲哚1m(收率61%)。1h nmr(300mhz,cdcl3)δ7.76(s,
1h),7.39(s,1h),7.30-7.13(m,5h),7.02(d,j=2.2hz,1h),6.94(dd,j=8.5,1.6hz,1h),6.84(d,j=8.5hz,1h),1.72(s,6h).
13
c nmr(75mhz,cdcl3)δ149.6,138.0,128.2,126.4,125.8,125.0,122.5,122.3,121.3,115.4,114.1,38.9,30.7.
[0169][0170]
通过实施例38得到无色油状吲哚1n(收率74%)。1h nmr(400mhz,cdcl3)δ7.89(s,1h),7.80(d,j=7.8hz,1h),7.77-7.66(m,2h),7.63(d,j=8.6hz,1h),7.48-7.31(m,3h),7.07(s,1h),7.02(d,j=2.1hz,1h),6.90(d,j=8.2hz,1h),6.62(d,j=8.2hz,1h),2.35(s,3h),1.83(s,6h).
13
c nmr(100mhz,cdcl3)δ147.7,137.6,133.5,131.9,131.5,128.1,127.7,127.5,126.4,125.8,125.7,125.3,123.9,123.6,120.9,120.8,120.1,111.1,39.1,30.6,21.7.hrms(esi):calculated for c
22h22
n[m+h]
+
:300.1747;found:300.1736.
[0171]
实施例39:重氮化合物2f,2j,2k的制备
[0172][0173]
向250ml的圆底烧瓶中加入溶解在11ml乙腈中的2-氯-1,3-二甲基氯化咪唑啉(1.27g,7.5mmol,1.5equiv)。将上述混合物降温至-20℃,加入nan3(553.0mg,8.4mmol,1.7equiv)。搅拌30分钟后加入相应的萘酚衍生物(5.0mmol,1.0equiv),et3n(1.4ml,10.0mmol,2.0equiv)以及四氢呋喃(22ml)。反应2小时后,通过薄层色谱点板监测反应。待原料消耗完全后,加入水淬灭反应,用乙酸乙酯和水萃取,收集有机相用无硫酸钠干燥。旋蒸除去溶剂后经过柱层析法分离纯化得到相应的重氮化合物2f,2j,2k。
[0174][0175]
通过实施例39得到黄色固体重氮化合物2f(收率86%)。1h nmr(400mhz,cdcl3)δ7.82(s,1h),7.57-7.48(m,2h),7.34-7.21(m,2h).
13
c nmr(100mhz,cdcl3)δ173.8,138.1,130.5,129.9,129.4,126.1,125.3,124.5,119.3.hrms(esi):calculated for c
10
h6cln2o[m+h]
+
:205.0163;found:205.0159.
[0176][0177]
通过实施例39得到黄色固体重氮化合物2j(收率76%)。1h nmr(400mhz,cdcl3)δ8.35(s,1h),7.50(s,1h),7.44(d,j=8.2hz,1h),7.19(d,j=8.2hz,1h),3.95(s,3h),2.44
(s,3h).
13
c nmr(100mhz,cdcl3)δ176.0,165.5,145.4,135.0,133.4,131.8,126.6,125.9,123.9,119.4,79.8,52.6,21.0.hrms(esi):calculated for c
13h11
n2o3[m+h]
+
:243.0764;found:243.0764.
[0178][0179]
通过实施例39得到黄色固体重氮化合物2l(收率86%)。1h nmr(300mhz,cdcl3)δ8.44(s,1h),7.87(d,j=1.7hz,1h),7.86-7.80(m,1h),7.65-7.56(m,2h),7.53-7.45(m,2h),7.44-7.37(m,1h),7.34(d,j=8.3hz,1h),3.94(s,3h).
13
c nmr(75mhz,cdcl3)δ175.8,165.4,145.6,139.2,138.2,131.0,129.9,129.1,128.0,126.9,126.4,124.2,119.9,80.0,52.7.hrms(esi):calculated for c
18h13
n2o3[m+h]
+
:305.0921;found:305.0921.
[0180]
表1实施例1-29的产物3的结构表征数据
[0181]
[0182]
[0183]
[0184]
[0185]
[0186]
[0187]
[0188]
[0189]
[0190]
[0191]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1