一种苯并环丁烯官能化聚硅氧烷及其制备方法
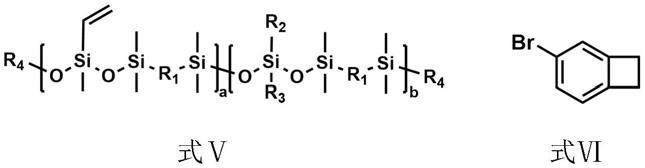
1.本发明属于高性能电子封装树脂领域,具体涉及一种苯并环丁烯官能化聚硅氧烷及其制备方法。
背景技术:
2.随着半导体和微电子工业的迅速发展,布线密度和集成度增加,随之产生的信号延迟和电路损耗成为阻碍其发展的瓶颈问题,低介电常数材料的研究成为一个重要课题。根据材料的成分特征,低介电材料主要可以分为无机材料和有机聚合物材料两大类。近些年来,有机聚合物因其介电常数低、平整度高、柔性可弯曲等优势,受到越来越多的关注。常见的聚合物基低介电常数聚合物材料主要包含聚四氟乙烯、聚酰亚胺、聚芳醚酮、聚对二甲苯、双马来酰亚胺树脂、苯并噁嗪树脂和苯并环丁烯树脂等,已经广泛应用于印刷电路板、平板显示和半导体封装的平坦化工艺和多层布线工艺中。其中,苯并环丁烯(benzocyclobutene,简称bcb)树脂具有优异的热、机械和介电性能,且在固化过程中无需催化剂,不会释放出小分子,在半导体封装的平坦化工艺和多层布线工艺中,作为一种的重要的芯片级封装的层间介质材料,拥有广阔的发展前景。
3.当前市场上商品化的bcb树脂是美国陶氏化学公司的cyclotene系列树脂,如美国专利5854302报道的方法,利用1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷(dvs)和4-溴苯并环丁烯(4-br-bcb)通过heck偶联制备双bcb官能团的dvs-bis-bcb单体,之后通过功能单体的溶液预聚合来控制树脂材料的分子量和分子量分布,以便适用于后续的晶圆级封装或多层布线工艺。该产品具有良好的热稳定性,低介电常数和介电损耗。
4.但这种制备方法存在明显的缺点:(1)多bcb官能团修饰的功能单体的制备需要多步化学反应和分离纯化步骤,提高了材料成本和开发门槛,如美国专利5136069、5354929的报道;(2)bcb基功能单体的开环聚合属于无规交联,需要严格控制预聚合条件,实时监测以避免体系交联度过高,甚至凝胶化的问题;(3)为了实现预聚物分子量的可控,bcb的开环聚合温度一般控制在160-180℃,属于引发bcb开环的起始温度区间,因此聚合速率较慢,导致预聚物的合成时间较长,整体材料制备费时费力。因此,目前急需探索简单高效的合成途径,方便调控bcb树脂材料的化学结构和性质,并降低生产成本,以适应日益增长的先进电子封装及低介电复合材料市场对不同性质要求的bcb树脂需求。
技术实现要素:
5.本发明的目的是提供一种苯并环丁烯官能化聚硅氧烷及其制备方法,该制备方法采用“预聚合-后修饰”方法,仅需两步反应,操作简单且产率可观。该苯并环丁烯官能化聚硅氧烷可热固化,固化后得到的材料热稳定性高,介电常数和介电损耗低。同时,该苯并环丁烯官能化聚硅氧烷可在紫外光下预固化形成目标图案,进一步加热完全固化。
6.第一方面,本发明保护式ⅰ所示苯并环丁烯官能化聚硅氧烷,
[0007][0008]
式ⅰ中,r1选自苯基或氧基;r2和r3各自独立地选自甲基、乙基、丙基、异丙基、正丁基、异丁基、辛基、十八烷基、环己基、苯基、三氟丙基、全氟辛基、乙烯基;r4表示三甲基硅基、二苯基甲基硅基或三苯基硅基;a为3~100中的任意整数;b为0~100中的任意整数。
[0009]
上述的苯并环丁烯官能化聚硅氧烷中,进一步地,a可为18~47中的任意整数;b可为0~42中的任意整数。
[0010]
所述苯并环丁烯官能化聚硅氧烷具体可为下述聚合物p1-p8中的任一种:
[0011]
聚合物p1:r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=18;b=42;
[0012]
聚合物p2:r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=18;b=42;
[0013]
聚合物p3:r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=28;b=28;
[0014]
聚合物p4:r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=28;b=28;
[0015]
聚合物p5:r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=37;b=16;
[0016]
聚合物p6:r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=37;b=16;
[0017]
聚合物p7:r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=47;b=0;
[0018]
聚合物p8:r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=47;b=0。
[0019]
第二方面,本发明保护所述的苯并环丁烯官能化聚硅氧烷的制备方法,包括如下步骤:
[0020]
(1)在惰性气氛下,式ⅱ所示化合物与式ⅳ所示化合物在催化剂ⅰ的作用下于有机溶剂ⅰ中进行缩合反应;或,
[0021]
在惰性气氛下,式ⅱ所示化合物与式ⅲ所示化合物和式ⅳ所示化合物在催化剂ⅰ的作用下于有机溶剂ⅰ中进行缩合反应;
[0022]
缩合完毕后加入封端剂进行封端,封端完毕后得到式
ⅴ
所示预聚物;
[0023][0024]
式ⅱ中,r1的定义同式ⅰ;
[0025]
式ⅲ中,r2和r3的定义同式ⅰ;r5选自甲基或乙基;
[0026]
式ⅳ中,r5选自甲基或乙基;
[0027]
(2)在惰性气氛下,式
ⅴ
所示预聚物与式ⅵ所示化合物(4-br-bcb)在缚酸剂、催化剂ⅱ和膦配体的作用下于有机溶剂ⅱ中进行偶联反应,得到式ⅰ所示苯并环丁烯官能化聚
硅氧烷;
[0028][0029]
式
ⅴ
中,r4的定义同式ⅰ。
[0030]
上述的制备方法中,以式ⅱ所示化合物的投料摩尔量为p,以式ⅲ所示化合物与式ⅳ所示化合物的投料摩尔量之和为q,以所述封端剂的投料摩尔量为r,p:q:r=1:(0.80~1.20):(0~0.5),如p:q:r=1:(0.80~1.20):0.07、1:1.0:0.07或1:0.95:0.07;
[0031]
式ⅲ所示化合物与式ⅳ所示化合物可取任意比例,如2.3:1、1:1。
[0032]
所述封端剂可为单烷氧基硅烷,优选为三甲基甲氧基硅烷、三甲基乙氧基硅烷、异丙氧基三甲基硅烷、三甲基丙氧基硅烷、二苯基甲基乙氧基硅烷、三苯基甲氧基硅烷或三苯基乙氧基硅烷;
[0033]
所述催化剂ⅰ与式ⅱ所示化合物的投料摩尔比可为0.001~0.1:1,具体可为0.01:1;
[0034]
所述催化剂ⅰ可为三(五氟苯基)硼;
[0035]
所述有机溶剂ⅰ可为超干甲苯、超干邻二甲苯、超干二氯甲烷和超干氯仿中的任意一种;
[0036]
所述缩合反应的温度可为0~60℃,具体可为室温(如25℃);时间可为0.5~24h,具体可为2h;
[0037]
所述封端的温度可为0~60℃,具体可为室温(如25℃);时间可为0.5~24h,具体可为0.5h。
[0038]
上述的制备方法中,式
ⅴ
所示预聚物与式ⅵ所示化合物的投料摩尔比可为1:(0.1~3),具体可为1:1;
[0039]
式ⅵ所示化合物与所述缚酸剂、所述催化剂ⅱ和所述膦配体的投料摩尔比为1:(1~10):(0.001~0.1):(0.003~0.3),具体可为1:(3.3~5.0):0.04:0.2、1:3.3:0.04:0.2、1:5:0.04:0.2或1:4.8:0.04:0.2;
[0040]
所述缚酸剂可为三乙胺、碳酸钾、碳酸钠和碳酸铯中的任意一种;
[0041]
所述催化剂ⅱ可为醋酸钯、四(三苯基膦)钯、二苯基膦二茂铁二氯化钯和钯碳中的任意一种;
[0042]
所述膦配体可为三苯基膦或者三(邻甲苯基)膦;
[0043]
所述偶联反应的温度可为25~110℃,具体可为回流温度;时间可为0.5~72h,具体可为12h。
[0044]
第三方面,本发明保护一种苯并环丁烯官能化聚硅氧烷固化物,由上述任一项所述的苯并环丁烯官能化聚硅氧烷在200~350℃固化而成。
[0045]
第四方面,本发明保护一种光刻胶,由上述任一项所述的苯并环丁烯官能化聚硅氧烷、光敏剂和有机溶剂组成。
[0046]
上述的光刻胶中各组分的质量份数如下:所述苯并环丁烯官能化聚硅氧烷1~1.5
份、光敏剂0.03~0.3份、有机溶剂5~50份。
[0047]
作为实施例,以质量份数计,所述光刻胶由1份所述苯并环丁烯官能化聚硅氧烷、0.1份光敏剂和8.7份有机溶剂组成。
[0048]
上述的光刻胶中,所述光敏剂可为式ⅶ所示氮烯类有机物,
[0049][0050][0051]
式ⅶ中,r表示任意基团,
[0052]
作为实例,所述光敏剂为式
ⅶ‑
1a所示化合物,但并不限于式
ⅶ‑
1a所示结构,
[0053][0054]
式
ⅶ‑
1a所示化合物的化学名称为1,12-双(4-(3-(三氟甲基)-3h-二嗪-3-基)苯基)十二烷-2,11-二酮;
[0055]
所述有机溶剂可为甲苯、邻二甲苯、均三甲苯、二氯甲烷、氯仿或四氢呋喃。
[0056]
本发明利用聚合物后修饰方法设计并合成了一系列苯并环丁烯官能化的聚硅氧烷,仅需两步反应,操作简单且产率可观。固化后得到的材料热稳定性高,介电常数和介电损耗低,其热力学性能和介电性能可根据主链和侧链的官能团的种类来调控。利用本发明聚合物和光敏剂复配的光刻胶固化后膜层的厚度均匀,图案边缘清晰。本体或改性后的树脂可以应用于大规模集成电路多芯片模块、高分子薄膜波导器、晶圆片级芯片规模封装、微电机系统、液晶显示器封装和信号绝缘组件封装等领域。
附图说明
[0057]
图1为本发明实施例5中制备得到的预聚物
ⅴ
的核磁氢谱图。
[0058]
图2为本发明实施例13中制备得到的聚合物p5的核磁氢谱图。
[0059]
图3为本发明实施例中聚合物p5固化后样品的热重分析曲线。
[0060]
图4为本发明实施例中聚合物p5固化后样品的介电常数随频率变化的谱图。
[0061]
图5为本发明实施例中聚合物p5和光敏剂复配光刻胶的光刻图案和轮廓仪测试曲线。
具体实施方式
[0062]
下面结合具体实施方式对本发明进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进的指南,并不以任何方式构成对本发明的限制。
[0063]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0064]
实施例1、预聚物1的制备
[0065]
在50ml三口瓶中依次加入磁子、1,4-二(二甲基硅烷基)苯3.888g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷0.754g(0.0057mol)、甲基苯基二甲氧基硅烷2.424g(0.0133mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率86.7%。本实施例制备得到的预聚物ⅰ中,r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=18;b=42。
[0066]
实施例2、预聚物ⅱ的制备
[0067]
在50ml三口瓶中依次加入磁子、1,1,3,3-四甲基二硅氧烷2.687g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷0.754g(0.0057mol)、甲基苯基二甲氧基硅烷2.424g(0.0133mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率50.3%。本实施例制备得到的预聚物ⅱ中,r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=18;b=42。
[0068]
实施例3、预聚物ⅲ的制备
[0069]
在50ml三口瓶中依次加入磁子、1,4-二(二甲基硅烷基)苯3.888g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷1.256g(0.0095mol)、甲基苯基二甲氧基硅烷1.732g(0.0095mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率96.1%。本实施例制备得到的预聚物ⅲ中,r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=28;b=28。
[0070]
实施例4、预聚物ⅳ的制备
[0071]
在50ml三口瓶中依次加入磁子、1,1,3,3-四甲基二硅氧烷2.687g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷1.256g(0.0095mol)、甲基苯基二甲氧基硅烷1.732g(0.0095mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗
过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率53.6%。本实施例制备得到的预聚物ⅳ中,r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=28;b=28。
[0072]
实施例5、预聚物
ⅴ
的制备
[0073]
在50ml三口瓶中依次加入磁子、1,4-二(二甲基硅烷基)苯3.888g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷1.759g(0.0133mol)、甲基苯基二甲氧基硅烷1.039g(0.0057mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率94.8%。本实施例制备得到的预聚物
ⅴ
中,r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=37;b=16。核磁氢谱图见图1。
[0074]
实施例6、预聚物ⅵ的制备
[0075]
在50ml三口瓶中依次加入磁子、1,1,3,3-四甲基二硅氧烷2.687g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷1.759g(0.0133mol)、甲基苯基二甲氧基硅烷1.039g(0.0057mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率51.9%。本实施例制备得到的预聚物ⅵ中,r1为氧基;r2为甲基;r3为苯基;r4为三甲基硅基;a=37;b=16。
[0076]
实施例7、预聚物ⅶ的制备
[0077]
在50ml三口瓶中依次加入磁子、1,4-二(二甲基硅烷基)苯3.888g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷2.512g(0.019mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率96.6%。本实施例制备得到的预聚物ⅶ中,r1为苯基;r2为甲基;r3为苯基;r4为三甲基硅基;a=47;b=0。
[0078]
实施例8、预聚物
ⅷ
的制备
[0079]
在50ml三口瓶中依次加入磁子、1,1,3,3-四甲基二硅氧烷2.687g(0.02mol)、三(五氟苯基)硼0.102g(0.0002mol)和超干邻二甲苯10ml,在氮气保护和冰浴的条件下滴加二甲氧基乙烯基甲基硅烷2.512g(0.019mol)和超干邻二甲苯10ml的混合物,滴加后室温下搅拌2小时,再加入过量甲氧基三甲基硅烷0.15g(0.0014mol),室温下反应0.5小时,最后加入中性氧化铝终止反应。粗产物通过漏斗过滤除去氧化铝,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次,旋蒸除去溶剂,最后得到不透明无色粘稠液体ⅰ,产率49.7%。本实施例制备得到的预聚物
ⅷ
中,r1为氧基;r2为甲基;r3为
6.214g(0.0339mol)、三乙胺24ml、色谱甲苯100ml、醋酸钯0.305g(0.0014mol)和三(邻甲基苯基)膦2.067g(0.0068mol),冰浴下氮气鼓泡30min。氮气保护,加热回流12h。粗产物过滤,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次。旋蒸除去溶剂,得到不透明浅黄色粘稠液体p7,产率85.1%。
[0094]
实施例16、苯并环丁烯修饰的聚合物p8的制备
[0095]
在250ml三口瓶中依次加入磁子、预聚物ⅰ10g(乙烯基0.0426mol)、4-br-bcb 7.807g(0.0426mol)、三乙胺30ml、色谱甲苯100ml、醋酸钯0.383g(0.0017mol)和三(邻甲基苯基)膦2.596g(0.0085mol),冰浴下氮气鼓泡30min。氮气保护,加热回流12h。粗产物过滤,旋干溶剂后,再溶解于10ml甲苯,加入100ml甲醇沉淀,静置,溶解并沉淀的过程重复三次。旋蒸除去溶剂,得到不透明浅黄色粘稠液体p8,产率84.9%。
[0096]
实施例17、苯并环丁烯修饰的聚硅氧烷的固化及性质表征
[0097]
将聚合物p5置于模具中,通过梯度升温(80℃/1h,150℃/1h,200℃/1h,235℃/1h,260℃/3h,300℃/1h)固化,得到浅黄色块材和片材。通过热重分析(图3)可知,固化后的p5的5%热失重温度在470℃以上。通过介电性能测试(图4)可知,固化后的p5的介电常数在2.5以下。其他聚合物的固化方法与实施例17一致。八种聚合物固化后的力学和介电性能如表1所示。
[0098]
表1不同化学结构树脂固化后的力学和介电性能对比
[0099][0100]
注:预聚物ⅰ~
ⅷ
中r4均代表三甲基硅基。
[0101]
实施例18、光刻胶的配制及光刻过程
[0102]
光敏剂1,12-双(4-(3-(三氟甲基)-3h-二嗪-3-基)苯基)十二烷-2,11-二酮的合成:在50ml三口瓶中加入癸二酰氯1.112g和二氯甲烷5ml,搅拌均匀后再将2.300g((4-(3-(三氟甲基)-3h-二氮杂萘-3-基)苯基)甲醇溶于10ml二氯甲烷,滴加至三口瓶中。室温下反应12h,旋蒸除去溶剂和多余单体,得到微黄色液体,产率95.6%。结构确认数据如下:1h-nmr(400mhz,cdcl3,δ/ppm):7.39-7.37(4h,d),7.20-7.18(4h,d),5.11(4h,s),2.36-2.32(4h,t),1.64-1.60(4h,m),1.30-1.27(8h,m)。
[0103]
将聚合物p5 100mg和光敏剂1,12-双(4-(3-(三氟甲基)-3h-二嗪-3-基)苯基)十二烷-2,11-二酮10mg溶于1ml甲苯溶液,搅拌均匀,配成光敏胶。将光敏胶旋涂在硅片上(1000rpm,10s;2000rpm,60s),80℃氮气烘箱烘烤5min,在波长365nm的紫外灯下曝光10s。
再置入烘箱110℃加热5min,显影液显影30s,再继续加热250℃固化1h。最终得到光刻图案,对其表面轮廓进行测试,轮廓仪测试曲线如图5所示,可以看出膜层的厚度均匀,光刻部分槽深130nm,曲线突变明显,说明光刻图案边缘清晰。
[0104]
以上对本发明进行了详述。对于本领域技术人员来说,在不脱离本发明的宗旨和范围,以及无需进行不必要的实验情况下,可在等同参数、浓度和条件下,在较宽范围内实施本发明。虽然本发明给出了特殊的实施例,应该理解为,可以对本发明作进一步的改进。总之,按本发明的原理,本技术欲包括任何变更、用途或对本发明的改进,包括脱离了本技术中已公开范围,而用本领域已知的常规技术进行的改变。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1