一种fap抑制剂和靶向fap的核素探针及应用
技术领域
1.本发明属于核医学药物领域,具体地,涉及一种fap抑制剂、基于该fap抑制剂的靶向fap的核素探针,以及所述抑制剂和探针在制备靶向fap的肿瘤显像剂/肿瘤治疗剂中的应用。
背景技术:2.成纤维细胞活化蛋白(fibroblast activation protein,fap),又称为脯氨酸内肽酶fap或seprase,是一种ⅱ型跨膜丝氨酸蛋白酶。在正常成人体内,fap在子宫内膜、子宫颈、胆囊、膀胱中存在一定程度的表达,在90%以上的上皮样恶性肿瘤间质中活化的成纤维细胞表面高表达,包括乳腺癌、结直肠癌、皮肤癌、前列腺癌和胰腺癌,但在其他组织、良性或癌前病变的上皮损伤中几乎不表达。此外,研究发现,fap在炎症和细胞纤维化相关的疾病中也明显表达,包括伤口愈合、类风湿性关节炎、骨关节炎、肝硬化、肺纤维化、心肌梗死后的心室重构。这些表达差异使得fap成为了癌症和炎症疾病潜在的影像学和放射治疗靶点。
3.经过近30年的发展,靶向fap的放射性诊疗药物取得了重大进展,包括放射性核素标记的抗体(如单克隆抗体mf19及其人源化产物sibrotuzumab、人鼠交叉反应抗体esc11和esc14、可结晶片段带有突变的抗体28h1)、环肽(如fap-2286)以及小分子抑制剂。其中,小分子抑制剂受到了最广泛的关注和研究,因其具有分子量低、组织穿透力强、特异性高、结构易于改造和修饰以便于放射性标记等特点。
4.目前,通过2-硼酸基、氰基或α-酮酰胺取代的吡咯烷来替代脯氨酸残基从而抑制fap催化中心的丝氨酸活性是fap小分子抑制剂的主要研发方向。其中,氰基类抑制剂受到了最广泛的关注,一方面,该类抑制剂对fap的选择性优于类似结构的硼酸类抑制剂,另一方面,其体内稳定性优于α-酮酰胺类抑制剂。
5.基于氰基类抑制剂的构效关系研究,jansen等人于2014年优选出n-(4-喹啉)-甘氨酸-(2-氰基-4,4-二氟)吡咯烷(uamc-1110)结构,该化合物具有高的fap亲和性和选择性(vs dpp iv、prep)、良好的体内稳定性和较为持久的fap抑制性(j med chem 2014,57(7),3053-3074)。随后,以uamc-1110为先导化合物开发了系列诊疗药物,已进行临床前研究的包括
68
ga-fapi-04、
68
ga-fapi-46、
68
ga-dota.sa.fapi、
68
ga-oncofap-dotaga、al
18
f-fapi-42、al
18
f-fapi-74、
99m
tc-fapi-34、
177
lu-fapi-04、
177
lu-dota.(sa.fapi)2等,为多种癌症病灶的精准定位和靶向杀伤提供了有力的工具。
68
ga-fapi-04、al
18
f-fapi-42用于心肌梗死、肺动脉高压患者的心脏活化的成纤维细胞成像研究提示,fap显像能够检测纤维化病灶。
6.177
lu-fapi-04等放射性治疗药物的肿瘤摄取较低、体内清除过快,限制了长半衰期治疗核素对靶细胞或组织的杀伤作用。因此,开发肿瘤摄取更高、滞留时间更长、非靶组织摄取低且清除快的fap靶向探针,对于推动fap靶向诊疗一体化药物具有重要意义。
技术实现要素:7.本发明的目的是提供一种fap抑制剂、基于该fap抑制剂的靶向fap的核素探针,以及所述抑制剂和探针在制备靶向fap的肿瘤显像剂/肿瘤治疗剂中的应用。
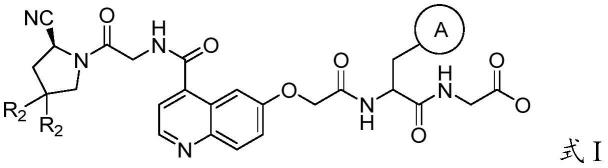
8.本发明的第一方面提供一种fap抑制剂,该fap抑制剂为具有式i所示结构的化合物中的至少一种,
[0009][0010]
其中,a为取代或未取代的苯基、吡啶基、吡唑基或噻吩基,r2为氢原子或卤素。
[0011]
根据本发明一种优选实施方式,a为苯基或吸电子基团取代的苯基,fap亲和性测试结果表明,苯环上加入吸电子基可进一步提高化合物的亲和性,所述吸电子基团优选为氰基、硝基、三卤甲基或卤素。
[0012]
本发明中,r2所代表的所述卤素优选为氟或氯,更优选为氟。
[0013]
更具体地,所述fap抑制剂选自以下化合物中的至少一种:
[0014]
[0015]
[0016][0017]
上述化合物可通过本领域常规有机合成方法制备得到,其可采用如图2所示的合成路线。路线中所涉及的各种原料均可商购获得,各步骤工艺条件均可参考有机合成领域公知的反应条件。
[0018]
本发明的第二方面提供一种靶向fap的核素探针,该探针为放射性核素标记的上述的fap抑制剂。通常,放射性核素标记在化合物羧基的一端。
[0019]
对探针进行放射性核素标记的方法可以为本领域常规方法。例如,首先在抑制剂上连接核素螯合基团,然后再标记放射性核素。
[0020]
上述方式获得的所述探针包括fap抑制剂单元和放射性核素标记单元,所述放射性核素标记单元包括核素螯合基团和放射性核素。
[0021]
根据本发明,所述核素螯合基团通常指双功能螯合剂形成的基团,所述双功能螯合剂可选自dota、nota、noda、nodaga、dotp、teta、atsm、ptsm、edta、ec、hbedcc、dtpa、sbad、bapen、df、dfo、tacn、no2a/notam、cb-do2a、cyclen、nota-aa、do3a、do3ap、hynic、mas3、mag3或异腈。
[0022]
上述双功能螯合剂的结构为本领域技术人员公知,例如,dota和nota结构分别如下所示:
[0023][0024]
根据目标用途不同,选择标记不同类型的放射性核素,例如,可标记诊断用放射性核素或治疗用放射性核素。
[0025]
根据本发明一种具体实施方式,所述探针的结构如配体cy01、配体cy02和配体cy03所示。
[0026]
本发明所述的fap抑制剂或靶向fap的核素探针可用于制备靶向fap的显像剂/治疗剂,包括肿瘤显像剂/肿瘤治疗剂。
[0027]
本发明的fap抑制剂表现出良好的亲和性,高于未修饰的化合物0,也高于文献报道的n-(4-喹啉)-2-氰基吡咯烷、uamc-1110。在显像实验中,由fap抑制剂进一步制备的放射性配合物在注射1h后,肿瘤区域即有明显的放射性浓集,除肾以外的非靶组织和器官摄取较低,注射2h后,肿瘤中仍滞留了一定量的放射性。与已被广泛接受的
68
ga-fapi-04相比,本发明的放射性配合物的肿瘤摄取和肿瘤/肌肉比值更高,具有良好的应用前景。
[0028]
本发明的其它特征和优点将在随后具体实施方式部分予以详细说明。
附图说明
[0029]
图1所示为化合物1-20的总结构式(1)和具体结构式(2)。
[0030]
图2所示为化合物1-20的制备总路线。
[0031]
图3所示为化合物27的制备路线。
[0032]
图4所示为化合物31的制备路线。
[0033]
图5-1至图5-18分别为化合物1至化合物5、化合物7至化合物13、化合物15至化合物20的hr-ms图,图5-19至图5-21分别为配体cy01、配体cy02和配体cy03的ms图。
[0034]
图6所示为配体cy01、cy02和cy03的合成步骤。
[0035]
图7所示为
68
ga-fapi-04、
68
ga-cy01和
68
ga-cy02在同一只荷u87mg肿瘤小鼠体内的pet显像结果(冠状位)。
[0036]
图8所示为
68
ga-cy01在荷瘤鼠体内抑制实验结果。
具体实施方式
[0037]
下面将更详细地描述本发明的优选实施方式。虽然以下描述了本发明的优选实施方式,然而应该理解,可以以各种形式实现本发明而不应被这里阐述的实施方式所限制。
[0038]
实施例
[0039]
小分子抑制剂的制备:
[0040]
化合物1-20的总结构式和具体结构如图1所示,合成步骤如图2所示。n'-芴甲氧羰基-甘氨酸-2-氯三苯基树脂(21,fmoc-gly-resin,取代容量为0.4mmol/g)购于上海楚肽生物科技公司,其他试剂购买于试剂公司,未经纯化。化合物27、化合物31由本实验室合成(如图3和图4所示)。氨基酸的偶联依照标准的fmoc固相合成法进行。
[0041]
化合物1-20的制备总路线。合成条件:(a)20%哌啶的dmf溶液,fmoc-x
1-oh、hbtu、hobt和dipea的dmf溶液;(b)20%哌啶的dmf溶液,化合物27/化合物31、hbtu、hobt和dipea的dmf溶液;(c)tfa,h2o。
[0042]
化合物27的制备路线。反应条件:(a)溴乙酸叔丁酯,cs2co3,dmf;(b)lioh,thf,h2o;(c)(s)-1-甘氨酰基吡咯烷-2-甲腈,hatu,dipea,超干dmf;(d)tfa,dcm。
[0043]
化合物24的合成:取6-羟基喹啉-4-羧酸(23,1g,5.29mmol),溴乙酸叔丁酯(2.27g,11.64mmol)和cs2co3(3.62g,11.11mmol)于50ml圆底烧瓶中,加入30ml dmf使之溶解,室温下搅拌,反应6小时后停止。减压除去dmf,向混合物中加入100ml h2o,ea萃取(50ml
×
3),收集有机相,经无水硫酸钠干燥后减压除去ea,粗产物经硅胶柱纯化后得黄色油状物
dose response模型,计算ic
50
。
[0051]
化合物的fap亲和性测试结果如表1所示。可以看到,r1基团中带有芳香基团的化合物相较于不含芳香基团的化合物(3和4)具有更高的亲和性;同时,苯环上加入吸电子基进一步提高了化合物的亲和性;由于空间位阻的关系,r1基团侧链上不能含有大体积的芳香环(如萘环)。当r1为l-(4-氰基)苯丙氨酸时,化合物18的亲和性相较于化合物0提高了26倍,也高于文献报道的n-(4-喹啉)-2-氰基吡咯烷(ic
50
:10.3
±
0.4nm;acs med.chem.lett.2013,4,491
–
496)、uamc-1110(ic
50
:3.2
±
0.4nm;j.med.chem.2014,57,3053
–
3074)的亲和性。亲和性的提高能够增加肿瘤摄取和延长滞留时间。
[0052][0053]
表1化合物的亲和性测试结果
[0054]
[0055][0056]
配体cy01、cy02、cy03的合成:
[0057]
配体cy01、cy02和cy03的合成步骤如图6所示。树脂(32,取代容量为0.4mmol/g)由本实验室制备,其他试剂购买于试剂公司,未经纯化。氨基酸的偶连依照标准的fmoc固相合
成法进行。
[0058]
树脂32的制备:取5g 2-ctc树脂于50ml固相合成管中,加二氯甲烷溶胀1小时,抽干溶剂后,加入n-fmoc-n
’‑
[1-(4,4-二甲基-2,6-二氧代环己亚基)乙基]-l-赖氨酸(2.13g)的dcm/dmf溶液(1:1,v/v)和3倍化学当量dipea,室温反应3小时。之后,使用dcm/meoh/dipea(10:10:1,v/v/v)封端四次,每次10分钟,经甲醇清洗后干燥至恒重,得到树脂32(负载量0.4mmol/g)。
[0059]
配体cy01、cy02和cy03的合成:取一定质量的树脂(32)于10ml固相合成管,加入2ml dcm溶胀,重复三次,每次5分钟,随后用dmf洗涤三次,每次5分钟。使用含20%哌啶的dmf溶液(v/v)脱去氨基保护基fmoc,具体操作为2ml 20%哌啶的dmf溶液反应2分钟、10分钟、10分钟,随后使用2ml dmf洗涤3-5次,每次2分钟。相对于树脂3倍化学量的氨基酸(fmoc-甘氨酸、fmoc-(4-氰基苯基)丙氨酸)在7.2倍化学量的dipea存在下,经3.6倍化学量的hbtu活化后加入到合成管中,电磁搅拌下反应1小时。化合物27、31、33和34的活化和偶连依上述方法进行。偶连化合物33或34前,使用含2%水合肼的dmf溶液(v/v)脱去氨基保护基dde,具体操作为2ml 2%水合肼的dmf溶液反应2分钟、3分钟、3分钟,随后使用2ml dmf洗涤3-5次,每次2分钟。化合物从树脂上解离和叔丁酯的脱去使用5ml tfa/h2o(95:5,v/v)搅拌2小时完成,并用2ml tfa清洗树脂,收集所有滤液,经减压除去tfa后,粗产物经hplc反相制备,冻干后得到目标产物cy01、cy02或cy03。化合物结构经低分辨质谱鉴定。图5-19至图5-21分别为配体cy01、配体cy02和配体cy03的ms图。
[0060]
标记与质控
[0061]
标记
[0062]
68
ga标记:精确称取一定质量配体于样品瓶中,加入适量dmso配制成10mm溶液。用移液枪吸取3μl配体溶液和65μl naoac溶液(1mol/l)于西林瓶中,加入1ml新淋洗得到的
68
ga
3+
离子溶液(溶剂为0.05mol/l的盐酸溶液,放射活度为37-74mbq),摇匀后密封,在45℃(配体为cy01或cy02)或95℃(配体为cy03)下反应15分钟。反应液冷却至室温后经tlc(薄层色谱法)分析质控。
[0063]
[
18
f]alf标记:用10ml生理盐水、10ml纯水依次淋洗sep-pak qma小柱,空气吹干备用。使用纯水将氟(
18
f)离子稀释至10ml,加到qma小柱中,以10ml纯水淋洗,空气吹干。取0.2ml生理盐水淋洗qma柱,前0.1ml弃去,收取后0.1ml待用。取20μl 0.5mol/l的khp、7μl 20mmol/l的alcl3溶液和100μl氟化钠加入到标记前体cy02(200μg,提前分装后冻干)瓶中,摇匀后室温放置5min,再加100μl乙醇于反应瓶中,混匀后110℃反应15min,不时摇动。待反应液冷却后,加入2ml纯水至反应瓶中,将溶液加入活化后的sep-pak vac c-18柱中,以3.0ml纯水淋洗杂质并弃去。加上0.2μm无菌微孔滤膜,以0.6ml 80%的乙醇溶液收集产品到无菌真空瓶中待用。取出适量淋洗液,使用hplc测定配合物的放射化学纯度。
[0064]
质控:
68
ga配合物的放射化学纯度使用tlc测定,固定相为快速硅胶纸,展开剂为饱和edta溶液,
68
ga
3+
在该体系下的rf值为0-9-1.0,标记产物
68
ga-cy01、
68
ga-cy02或
68
ga-cy03的rf值均为0-0.1。[
18
f]alf配合物的放射化学纯度使用hplc(高效液相色谱)测定,流动相为含0.1%三氟乙酸/水、0.1%三氟乙酸/乙腈,终产物[
18
f]alf-cy02的保留时间为8.0min。配合物的放射化学纯度均大于90%,未经进一步纯化即进行下一步研究。
[0065]
68
ga标记产物的显像
[0066]
取0.1ml新制备的
68
ga标记配合物(5.6mbq-7.4mbq)经尾静脉注射到荷u87mg肿瘤的雄性balb/c裸鼠体内。于注射1h后用异氟烷麻醉,进行小动物pet/ct(super-nova,平生科技,中国)显像,勾画感兴趣区的标准摄取值suv
max
。在
68
ga-cy01的抑制实验中,提前半小时经小鼠尾静脉注射100nmol未标记配体cy01,注射放射性配合物1h后对其进行pet/ct显像。
[0067]
如图7和表2所示,注射放射性配合物1h后,肿瘤区域即有明显的放射性浓集,除肾以外的非靶组织和器官摄取较低。与已被广泛接受的
68
ga-fapi-04相比,在同一只荷瘤鼠体内(第一天做
68
ga-fapi-04显像,第3天上午
68
ga-cy02做显像,下午
68
ga-cy01做显像),注射1h后,
68
ga-cy01和
68
ga-cy02的肿瘤摄取和肿瘤/肌肉比值更高,肿瘤suvmax分别是
68
ga-fapi-04的3.6和4.3倍,肿瘤/肌肉比值分别是
68
ga-fapi-04的2.2和2.0倍。在抑制实验中(如图8所示),cy01能够明显抑制
68
ga-cy01在肿瘤部位的浓集,表明
68
ga-cy01与fap蛋白进行了特异性结合。
[0068]
表2配合物在肿瘤、肌肉和肾脏中suvmax值及肿瘤/肌肉比值(mean
±
sd,n=3)
[0069][0070]
以上已经描述了本发明的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。