一种聚合度可控的壳寡糖化学合成方法
1.本发明涉及一种聚合度可控的壳寡糖化学合成方法,属于糖化学领域。
背景技术:
2.壳寡糖是由2~20个氨基葡萄糖或乙酰氨基葡萄糖以β-1,4糖苷键连接而成的低聚糖,广泛应用于生物医药、日用化工、农业、食品等领域。与大分子的壳聚糖相比,壳寡糖因具有低粘度、高水溶性、高生物相容性以及可生物降解性等良好的性质而受到关注。壳寡糖具有许多生理活性,包括抗菌、抗肿瘤、抗氧化、免疫调节、血糖调节等。壳寡糖的细胞毒性小,容易被人体小肠吸收和利用。不同的聚合度和乙酰化程度直接影响其生物活性。
3.近年来,壳寡糖的制备大多是通过提取甲壳类动物外骨骼中的甲壳素,经脱乙酰和水解作用制备得到。甲壳素的提取可归纳为“三脱”,分别是脱蛋白质、脱盐、脱色,需要对甲壳素进行酸、碱和还原剂的长时间处理。通常情况下去乙酰化通过与高浓度碱液作用完成,糖链的降解包括酸解法、氧化降解法、硼酸钠降解法、超声波法、微波法、光降解法和酶法。但是,通过降解壳聚糖制备的壳寡糖会有一定程度上的异质源的残留,如蛋白质、盐等。这为后续纯化造成了一定的困难。
4.随着近几年糖化学的兴起,寡糖的化学合成成为一个热点,通过化学法可以控制壳寡糖合成的聚合度,满足其在不同领域的要求。
技术实现要素:
5.技术问题:本发明所要解决的技术问题是合成不同聚合度的壳寡糖(β-1,4-寡聚-葡萄糖胺),并通过改变糖砌块的保护基策略和糖苷化反应条件实现聚合度可控。在此之前,只需设计合成异头碳上连有可活化的离去基团并含有c-4裸露羟基的糖砌块即可。
6.技术方案:
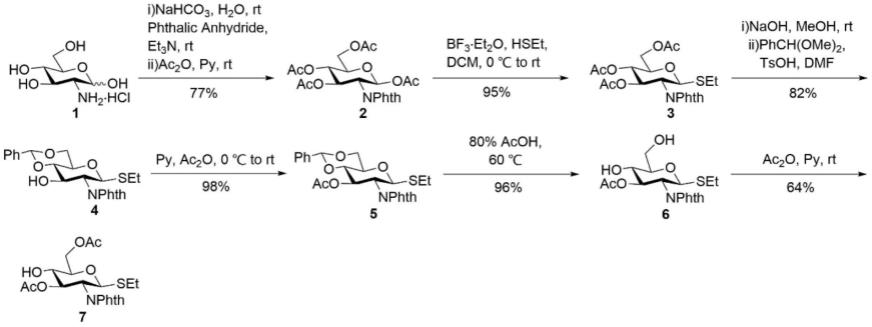
7.本发明提供了一种聚合度可控的壳寡糖的化学合成方法,壳寡糖是由2~20个葡萄糖胺通过β-1,4糖苷键连接而成,该合成方法包括如下过程:
8.(1)利用式i所示的单糖砌块作为底物,在促进剂的作用下发生自组装糖苷化反应:一个糖砌块异头碳的离去基团被活化之后,连接另一个糖砌块的c-4羟基,生成不同聚合度的全保护的壳寡糖;
[0009][0010]
其中,r选自乙硫基(set),对甲苯硫基(stol),苯硫基(sph),三氟乙酰亚胺酯基(ocnphcf3),二苄氧磷酸酯基(op(obn)2)等能够被活化的离去基团;r1选自邻苯二甲酰基(phth),三氯乙氧基羰基(troc),苄甲氧羰基(cbz)等氨基保护基;r2、r3分别独立选自乙酰基(ac),苯甲酰基(bz),乙酰丙酰基(lev),苄基(bn)或萘亚甲基(nap)等保护基;r2、r3不同
时为苄基;
[0011]
(2)进行全脱保护反应,得到壳寡糖产物。
[0012]
在本发明的一种实施方式中,式i所示的单糖砌块中,它含有在异头碳可被活化的离去基团,c-4含有一个裸露的羟基,使其既作为供体,又作为受体;对于异头碳,一些可被活化的离去基团(r)可被选用,比如常见的硫糖苷、磷酸酯基糖苷、糖基亚砜、亚胺酯基糖苷等;对于氨基保护基(r1),可选用的是邻苯二甲酰基(phth)、三氯乙氧羰基(troc)或苄氧羰基(cbz);对于c-3、c-6羟基的保护基(r2、r3)可分别使用乙酰基、苄基、2-萘亚甲基、对甲氧基苄基进行保护。
[0013]
在本发明的一种实施方式中,r3优选乙酰基(ac),苯甲酰基(bz),乙酰丙酰基(lev),萘亚甲基(nap)。
[0014]
在本发明的一种实施方式中,通过改变糖砌块保护基、反应时间、浓度以及不同的促进剂来改变寡糖聚合度。糖砌块保护基不同时,影响了糖砌块的糖基化反应活性,进而影响了连接的聚合度。而且当糖砌块保护基供电子能力较强时,会导致反应活性过高而产生大量的副产物,无法得到要求聚合度的壳寡糖。
[0015]
在本发明的一种实施方式中,所述促进剂为nis和tfoh。在本发明中,促进剂可以为nis、tfoh、meotf中的一种或两种,通过试验证明,nis和tfoh共同作为促进剂时,自组装糖苷化反应转化率最高,比单独使用任意一种具有显著的优势。
[0016]
在本发明的一种实施方式中,所述自组装糖苷化反应是在有机溶剂中进行的,自组装糖苷化反应中底物浓度为0.02~0.5m。所述有机溶剂优选为二氯甲烷(dcm)。
[0017]
进一步地,在本发明的一种实施方式中,所述自组装糖苷化反应中,有机溶剂的用量为:12-15ml/mmol单糖砌块。
[0018]
在本发明的一种实施方式中,所述自组装糖苷化反应中,还包括加入分子筛。分子筛的加入是为了尽可能除去反应溶剂中的水,减少副反应的发生。
[0019]
在本发明的一种实施方式中,所述自组装糖苷化反应中,nis与单糖砌块的摩尔比为1.5-3.0:1;tfoh与单糖砌块的摩尔比为0.1-0.3:1。
[0020]
进一步地,在本发明的一种实施方式中,所述自组装糖苷化反应中,nis与单糖砌块的摩尔比为1.8-2.5:1;tfoh与单糖砌块的摩尔比为0.15-0.25:1。
[0021]
在本发明的一种实施方式中,所述自组装糖苷化反应中,温度控制在-78℃,时间为1-5h。
[0022]
在本发明的一种实施方式中,自组装糖苷化反应包括如下过程:将糖砌块溶于dcm中,加入分子筛,将混合物冷却至-78℃;加入nis和tfoh,反应4h后;反应结束后,加入三乙胺淬灭反应,反应液经dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩,柱层析分离纯化。
[0023]
在本发明的一种实施方式中,当r为乙硫基(set),r1为邻苯二甲酰基(phth),r2为乙酰基(ac)、r3为乙酰基(ac)时,相应单糖砌块通过如下合成路线制得:
[0024][0025]
以化合物1葡萄糖胺盐酸盐为起始原料,在碱的作用下去除盐酸,并加入弱碱在c-2位上氨基保护基,所得化合物在吡啶和乙酸酐作用下全乙酰化,得到化合物2;
[0026]
化合物2在路易斯酸的作用下与乙硫醇生成硫苷化合物3;
[0027]
所得硫苷化合物3在碱性条件下脱除乙酰基后与苯甲醛二甲缩醛在对甲苯磺酸的催化下生成4,6-苄叉基保护的化合物4;
[0028]
而后把3-oh用乙酰基保护后得到全保护化合物5;
[0029]
化合物5在醋酸水溶液中脱除苄叉基,得到化合物6;
[0030]
化合物6在吡啶作用下与乙酸酐反应生成单糖砌块7。
[0031]
在本发明的一种实施方式中,当r为乙硫基(set),r1为邻苯二甲酰基(phth),r2为苄基(bn)、r3为乙酰基(ac)时,相应单糖砌块通过如下合成路线制得:
[0032][0033]
化合物4在三号位上苄基化得到8,在酸性条件下打开4,6-苄叉基得到9,最后在六号位选择性上乙酰基得到单糖砌块10。
[0034]
在本发明的一种实施方式中,自组装糖苷化反应合成可控聚合度壳寡糖的过程如下所示:
[0035]
n为2~20。
[0036]
在本发明的一种实施方式中,全脱保护后所得壳寡糖产物的结构如下所示:
[0037]
n为2~20。
[0038]
在本发明的一种实施方式中,n具体可为2~6,或者2~3。
[0039]
在本发明的一种实施方式中,通过对全保护的壳寡糖的进行全脱保护反应,得到不同聚合度的壳寡糖。
[0040]
其中,基和邻苯二甲酰基在水合肼的作用下脱除;苄基在钯碳的作用下脱除;其它保护基可采用现有方法脱除。
[0041]
有益效果:
[0042]
本发明可通过化学合成制备不同聚合度的壳寡糖,并通过改变糖砌块的保护基策略和糖苷化反应条件实现聚合度可控。
附图说明
[0043]
图1为壳寡糖a的质谱表征。
[0044]
图2为壳寡糖a的hplc分析。
[0045]
图3为壳寡糖b的质谱表征。
[0046]
图4为壳寡糖b的hplc分析。
[0047]
图5为壳寡糖c的质谱表征。
[0048]
图6为壳寡糖c的hplc分析。
[0049]
图7为壳寡糖d的质谱表征。
[0050]
图8为壳寡糖e的质谱表征。
[0051]
图9为壳寡糖f的质谱表征。
具体实施方式
[0052]
以下通过实施例来进一步描述本发明的有益效果,应理解为,这些实施例仅用于例证的目的,决不限制本发明的范围。实施例中未注明具体条件者,按照常规条件进行。所用试剂或仪器未注明生产商者,均为可以通过市购获得的常规产品。
[0053]
本发明的产率的计算方法为“产物(mol)/反应底物(mol)*100%”。本发明中化合物的核磁数据由bruker ascend 400m/600m核磁共振仪在25℃下测得;质谱数据由tsq quantum ultra emr测定;旋光度由schmit&ha nsch unipo l1000全自动旋光仪在589nm下测得,测定浓度(c)单位为g/100ml。
[0054]
实施例1:
[0055]
糖砌块7的合成路线如下:
[0056][0057]
以葡萄糖胺盐酸盐1为起始原料,在碱的作用下去除盐酸,并加入弱碱在c-2位上
d)δ7.98
–
7.70(m,4h,ar),7.53
–
7.33(m,5h,ar),5.92(t,j=9.4hz,1h,h-3),5.58(d,j=10.6hz,1h,h-1),5.55(s,1h,arch),4.45
–
4.41(m,1h,h-5),4.37(dd,j=10.6,9.9hz,1h,h-2),3.86
–
3.75(m,3h,h-4,h-6a,h-6b),2.69(ttd,j=12.5,7.4,5.1hz,2h,set),1.90(s,3h,oac),1.21(t,j=7.4hz,3h,set).
13
c nmr(151mhz,chloroform-d)δ170.12,167.80,167.42,136.94,134.43,134.18,131.76,131.25,129.17,128.26,126.26,123.71,123.65,101.68,81.78,79.31,70.60,70.54,68.68,54.33,24.44,20.57,14.93.
[0063]
化合物6:将化合物5(3.26g,6.74mmol)溶解于80%乙酸水溶液(60.0ml)中,反应液在65℃下搅拌3h。浓缩后用柱层析纯化(二氯甲烷/甲醇,50:1),得到化合物6(2.55g,96%)。[α]
22d
=4.27(c 1.0,chcl3).1h nmr(400mhz,chloroform-d)δ7.97
–
7.66(m,4h,ar),5.72(dd,j=10.2,8.9hz,1h,h-3),5.55(d,j=10.5hz,1h,h-1),4.30(t,j=10.4hz,1h,h-2),4.01(dd,j=12.1,3.3hz,1h,h-6a),3.90(dd,j=12.0,4.7hz,1h,h-6b),3.81(t,j=9.4hz,1h,h-4),3.71(ddd,j=9.8,4.6,3.2hz,1h,h-5),2.69(qq,j=12.6,7.4hz,2h,set),1.94(s,3h,oac),1.21(t,j=7.4hz,3h,set).
13
c nmr(151mhz,chloroform-d)δ171.42,167.96,167.42,134.47,134.27,131.68,131.24,123.69,81.10,79.76,74.55,70.21,62.51,53.82,24.41,20.70,14.97.
[0064]
化合物7:将化合物6(2.35g,5.94mmol)溶解于吡啶(15.0ml)中,在0℃下加入乙酸酐(0.84ml,8.91mmol),在0℃下搅拌12h,反应液用饱和碳酸氢钠溶液洗涤。dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(石油醚/乙酸乙酯,2:1),得到化合物7(1.65g,64%)。[α]
22d
=-7.34(c 1.0,chcl3).1h nmr(400mhz,chloroform-d)δ7.81(ddd,4h,ar),5.70(dd,j=10.3,8.9hz,1h,h-3),5.50(d,j=10.5hz,1h,h-1),4.53
–
4.45(m,1h,h-6a),4.41(dd,j=12.2,2.3hz,1h,h-6b),4.31(t,j=10.4hz,1h,h-2),3.78(ddd,j=10.0,4.7,2.3hz,1h,h-5),3.66(dd,j=10.0,8.9hz,1h,h-4),2.79
–
2.59(m,2h,set),2.14(s,3h,oac),1.93(s,3h,oac),1.22(t,j=7.4hz,3h,set).
13
c nmr(101mhz,chloroform-d)δ171.70,171.23,167.87,167.42,134.45,134.28,131.65,131.23,123.69,81.20,77.36,74.08,69.63,63.31,53.67,24.52,20.91,20.69,14.94.
[0065]
实施例2
[0066]
糖砌块10的合成如如下:
[0067][0068]
化合物4在三号位上苄基得到8,在酸性条件下打开4,6-苄叉基得到9,最后在六号位选择性上乙酰基得到化合物10。
[0069]
具体实验操作和步骤:
[0070]
化合物8:将化合物4(5.27g,11.5mmol)溶于dmf(40.0ml)中,在0℃下,加入60%氢化钠(688mg,17.2mmol)并搅拌0.5h,向反应物中加入溴苄(2.04ml,17.2mmol)。在常温下反应4h,待到反应物完全消失,在0℃下加入水淬灭反应,二氯甲烷萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(石油醚/乙酸乙酯,10:1),得到化合物8(5.3g,85%)。
[α]
22d
=43.18(c 1.0,chcl3).1h nmr(400mhz,chloroform-d)δ7.91
–
7.65(m,4h,ar),7.61
–
7.37(m,5h,ar),7.05
–
6.89(m,5h,ar),5.66(s,1h,arch),5.38(d,j=10.6hz,1h,h-1),4.83(d,j=12.3hz,1h,arch),4.54(d,j=12.3hz,1h,arch),4.49(dd,j=10.0,8.9hz,1h,h-3),4.44(dd,j=10.3,4.8hz,1h,h-6a),4.38-4.28(m,1h,h-2),3.87(dd,j=9.7,5.3hz,1h,h-6b),3.83(d,j=3.6hz,1h,h-4),3.73(td,j=9.8,4.9hz,1h,h-5),2.68(qq,j=12.5,7.5hz,2h,set),1.19(t,j=7.4hz,3h,set).
[0071]
化合物9:将化合物8(5.00g,9.10mmol)溶解于80%乙酸水溶液(90.0ml)中,反应液在65℃下搅拌5h。浓缩后用柱层析纯化(二氯甲烷/甲醇,50:1),得到化合物9(3.7g,100%)。[α]
22d
=34.67(c 1.0,chcl3).1h nmr(400mhz,chloroform-d)δ7.86
–
7.67(m,4h,ar),7.13
–
6.94(m,5h,ar),5.31(d,j=10.3hz,1h,h-1),4.71(d,j=12.2hz,1h,arch),4.55(d,j=12.1hz,1h,arch),4.31(dd,j=10.2,8.5hz,1h,h-3),4.23(t,j=10.2hz,1h,h-2),3.96(dd,j=11.9,3.5hz,1h,h-6a),3.87(dd,j=12.0,4.6hz,1h,h-6b),3.80(dd,j=9.8,8.5hz,1h,h-4),3.58(ddd,j=9.6,4.5,3.4hz,1h,h-5),2.64(ttd,j=12.6,7.5,5.1hz,2h,set),1.17(t,j=7.4hz,3h,set).
13
c nmr(101mhz,chloroform-d)δ168.24,167.50,137.93,134.05,133.97,131.60,128.34,128.27,127.85,127.68,123.65,123.36,81.42,80.31,79.37,74.65,72.23,62.67,54.61,24.22,14.93.
[0072]
化合物10:将化合物9(2.58g,5.60mmol)溶解于吡啶(18.6ml)中,在0℃下加入乙酸酐(0.53ml,5.60mmol),在0℃下搅拌10h,反应液用饱和碳酸氢钠溶液洗涤。dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(石油醚/乙酸乙酯,4:1),得到化合物10(1.4g,51%)。[α]
22d
=17.01(c 1.0,chcl3).1h nmr(400mhz,chloroform-d)δ7.87
–
7.65(m,4h,ar),7.08
–
6.93(m,5h,ar),5.28(d,j=10.1hz,1h,h-1),4.74(d,j=12.1hz,1h,arch),4.55(d,j=12.1hz,2h,arch,h-6a),4.32(dd,j=12.2,1.8hz,1h,h-6b),4.30
–
4.26(m,1h,h-3),4.22(t,j=10.1hz,1h,h-2),3.64(d,j=2.1hz,2h,h-4,h-5),2.74
–
2.53(m,2h,set),2.14(s,3h,oac),1.18(t,j=7.4hz,3h,set).
13
c nmr(151mhz,chloroform-d)δ172.01,168.14,167.51,137.93,134.02,133.92,131.60,128.25,127.92,127.59,123.61,123.33,81.41,79.51,78.05,74.72,71.68,63.37,54.54,24.23,20.91,14.89.
[0073]
实施例3:壳寡糖衍生物的制备
[0074]
通过对两种不同糖砌块的自组装糖苷化,形成了两种不同壳寡糖衍生物的混合物。本实例采用两种不同的硫苷,氨基保护基采用邻苯二甲酰基,c-4,c-6分别用乙酰基或苄基保护。对合成的壳寡糖衍生物通过maldi-tof,hplc进行表征。
[0075]
糖苷化一般步骤如下式所示:
[0076][0077]
具体实验操作和步骤:
[0078]
将糖砌块7(108mg,0.25mmol)溶于dcm(3.10ml),加入分子筛,将混合物冷却至-78℃。向反应液中加入nis(111mg,0.49mmol)和tfoh(4μl,0.05mmol),反应4h后,加入三乙胺淬灭反应,反应液经dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(二氯甲烷/甲醇,50:1),得到混合物a(86mg);壳寡糖a的
质谱表征数据如图1所示,hplc分析数据如图2所示。
[0079]
将糖砌块10(98mg,0.2mmol)溶于dcm(2.5ml)。加入分子筛,将混合物冷却至-78℃,向反应液中加入nis(90.8mg,0.40mmol)和tfoh(3.2μl,0.04mmol),反应4h后,加入三乙胺淬灭反应,dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(二氯甲烷/甲醇,50:1),得到混合物b(67mg);壳寡糖b的质谱表征数据如图3所示,hplc分析数据如图4所示。
[0080]
将糖砌块7(113mg,0.26mmol)溶于dcm(3.20ml),加入分子筛,将混合物冷却至-15℃,向反应液中加入meotf(80μl,0.78mmol),反应48h后,加入三乙胺淬灭反应,dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(二氯甲烷/甲醇,50:1),得到混合物c(66mg);壳寡糖c的质谱表征数据如图5所示,hplc分析数据如图6所示。
[0081]
将糖砌块7(54mg,0.12mmol)溶于dcm(1.50ml),加入分子筛,将混合物冷却至-78℃。向反应液中加入nis(55mg,0.25mmol)和tmsotf(4.8μl,0.02mmol),反应4h后,加入三乙胺淬灭反应,反应液经dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(二氯甲烷/甲醇,50:1),得到混合物d(44mg);壳寡糖d的质谱表征数据如图7所示。
[0082]
合成结果:
[0083]
糖砌块7在nis和tfoh作用下自组装连接所得寡糖产物a的质谱分析和hplc分析如图1、图2所示(需说明的是,在自组装糖苷化后形成的壳寡糖衍生物,还原端异头碳会出现水解,这是由于硫苷被活化之后不能再回到初始状态,但并不影响实验进行与后续脱保护)。通过hplc和质谱分析,其中,二糖4%,三糖11%,四糖30%,五糖24%,六糖19%,七糖7%,八糖2%,九糖1%。
[0084]
糖砌块10在nis和tfoh作用下自组装连接所得寡糖产物b的质谱分析和hplc分析如图3、图4所示。通过hplc以及质谱分析,其中,二糖44%,三糖20%,四糖9%,五糖19%,六糖5%。
[0085]
糖砌块7在meotf作用下自组装连接所得寡糖产物c的质谱分析和hplc分析如图5、图6所示。通过hplc以及质谱分析,其中,二糖11%,三糖16%,四糖56%,五糖12%,六糖1%。
[0086]
糖砌块7在nis和tmsotf作用下自组装连接所得寡糖产物d的质谱分析图7所示。通过对产物纯化后得出其中单糖60%,二糖35%,三糖5%。
[0087]
具体结果见表1。
[0088]
表1不同糖砌块、不同条件对聚合度的影响
[0089][0090][0091]
实施例4:全脱保制备壳寡糖
[0092]
将壳寡糖a溶于溶于甲醇中(21mg)将壳寡糖a(21mg)溶于甲醇中(1.0ml),加入水合肼(1.0ml),在65℃下回流反应两天,浓缩过滤,用c18反向柱纯化得到全脱保壳寡糖a’(6.5mg)。(壳寡糖a,在水合肼的条件可同时脱去乙酰基和邻苯二甲酰基)。
[0093][0094]
将壳寡糖b(28mg)溶于dcm/t-butanol中(1:1,v/v,1.0ml),并加入钯碳(pd/c)后搅拌,混合物在1个大气压下的h2压力下反应三天,浓缩过滤,得到半脱保产物。半脱保产物溶于甲醇中(1.0ml),加入水合肼(1.0ml),在65℃下回流反应两天,浓缩过滤,用c18反向柱纯化得到全脱保壳寡糖b’(5.9mg)
[0095][0096]
对比例1
[0097]
糖砌块11的合成路线如下所示:
[0098][0099]
具体操作如下:
[0100]
氩气保护下,将化合物8(3.55g,6.68mmol)溶于dcm中(70ml),在0℃下,先后滴加三乙基硅烷(12.8ml,80mmol)和三氟化硼乙醚(1.68ml,13.3mmol),在室温下搅拌3h,待到反应物完全消失,加入三乙胺淬灭反应,所得反应液用饱和碳酸氢钠水溶液洗涤,二氯甲烷萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(石油醚/乙酸乙酯,5:1),得到
化合物11(2.9g,88%)。[α]
22d
=+32.38(c 1.0,ch3cl).1h nmr(400mhz,chloroform-d)δ7.87
–
7.64(m,4h,ar),7.43
–
7.31(m,5h,ar),7.13
–
6.92(m,5h,ar),5.27(d,j=10.0hz,1h,h-1),4.75(d,j=12.2hz,1h,arch),4.64(d,j=11.9hz,1h,arch),4.60(s,1h,arch),4.54(d,j=12.1hz,1h,arch),4.29(d,j=10.2hz,1h,h-2),4.28
–
4.18(m,1h,h-3),4.03
–
3.83(m,1h,h-6a),3.84(d,j=4.9hz,1h,h-4),3.77(dd,j=10.1,5.2hz,1h,h-6b),3.68(dt,j=9.8,5.0hz,1h,h-5),2.73
–
2.52(m,2h,set),1.16(t,j=7.4hz,3h,set).
13
c nmr(151mhz,chloroform-d)δ168.10,167.55,138.14,137.60,133.94,133.85,131.68,128.55,128.19,127.93,127.84,127.47,123.56,123.30,81.20,79.60,74.57,74.49,73.83,70.94,54.42,23.99,14.92.
[0101]
将糖砌块11(134mg,0.25mmol)溶于dcm(3.10ml),加入分子筛,将混合物冷却至-78℃。向反应液中加入nis(111mg,0.49mmol)和tfoh(4μl,0.05mmol),反应4h后,加入三乙胺淬灭反应,反应液经dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(二氯甲烷/甲醇,50:1),得到混合物e;壳寡糖e的质谱表征数据如图8所示,所得产物组分分子量与预期分子量不符,说明3,6-二苄基葡萄糖砌块并不适用在此方法下进行糖基化反应。
[0102]
将糖砌块11(220mg,0.41mmol)溶于dcm(5.10ml),加入分子筛,将混合物冷却至-15℃,向反应液中加入meotf(128μl,1.24mmol),反应48h后,加入三乙胺淬灭反应,dcm稀释后用饱和碳酸氢钠洗涤,dcm萃取三次,有机相用无水na2so4干燥,浓缩。柱层析分离纯化(二氯甲烷/甲醇,50:1),得到混合物f(188mg)。质谱分析结果如图9所示,由图9可知3,6-二苄基葡萄糖砌块同样也无法有效进行糖基化反应合成壳聚糖产物。
[0103]
通过对比例1与实施例4(表1)的比较,说明由于苄基较强的供电子效应,提高了4,6-二苄基保护的糖砌块11的活性,导致糖砌块在自组装反应过程中过于活泼而产生大量的副产物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1