一种精准定量调控大肠杆菌重组蛋白胞外分泌水平的方法及应用
1.本发明涉及一种精准定量调控大肠杆菌重组蛋白胞外分泌水平的方法及应用,属于基因工程与发酵工程领域。
背景技术:
2.大肠杆菌表达系统是目前基因工程中最常用的重组蛋白表达系统具有遗传背景清楚,目的基因表达水平高,培养周期短,抗污染能力强等特点。大肠杆菌在基因表达技术中占有重要的地位,是分子生物学研究和生物技术产业化发展进程中的重要工具。是常用的重组蛋白高效生产的宿主之一,但是在大肠杆菌的异源表达中,大部分重组蛋白在信号肽的引导下分泌到周质空间,会导致中间体大量积聚,对蛋白质的生产造成阻碍。
3.肽聚糖是由双糖单位,四肽尾还有肽桥聚合而成得多层网状大分子结构。肽聚糖层的骨架由n-乙酰葡萄糖胺和n-乙酰胞壁酸通过β-1,4糖苷键连接而成,糖链间由肽链交联,构成稳定的网状结构,这样构成了整个细菌表面的细胞壁。作为细胞壁的主要成分,肽聚糖的组成与细胞结构的完整性、外膜的通透性的维持、细菌的抗干燥能力和耐热性等均密切相关。cn105755029a公开了一种基于daca提高大肠杆菌重组蛋白胞外分泌水平的方法,但是d,d-羧肽酶自身对菌体具有毒性的缺点。因此,该方法提供一种可以解决该问题的方法,对提高重组蛋白的胞外分泌的进一步研究具有重要价值。
技术实现要素:
4.本发明提供了一种重组质粒,含有第一启动子、第二启动子、色氨酸合成酶基因簇trpedcba、操纵区trpo和d,d-羧肽酶基因daca;所述操纵区trpo具有可与由阻遏蛋白trpr和色氨酸形成的共价二聚体结合的结合位点;所述第一启动子调控trpedcba的表达;所述第二启动子调控操纵区trpo的基因表达;所述操纵区trpo的下游连接d,d-羧肽酶基因daca;所述第一启动子和第二启动的转录方向相反。
5.在一种实施方式中,所述第一启动子为t7启动子,核苷酸序列如seq id no.6所示。
6.在一种实施方式中,所述第二启动子为在稳定期具有强起始转录能力的启动子,核苷酸序列如seq id no.7~11任一所示。
7.本发明还提供了色氨酸操纵子系统,包括敲除了色氨酸合成酶基因簇trpedcba的大肠杆菌细胞和所述重组质粒。
8.本发明还提供了色氨酸操纵子和温敏阻遏系统共表达的重组质粒,含有spp型启动子、低温诱导型启动子pr、高温诱导型启动子p
l
、色氨酸合成酶基因簇trpedcba、操纵区trpo、d,d-羧肽酶基因daca和温敏阻遏蛋白ci
ts
857;所述操纵区trpo具有可与由阻遏蛋白trpr和色氨酸形成的共价二聚体结合的结合位点;所述启动子p
l
上具有温敏阻遏蛋白ci
ts
857的结合位点;所述启动子p
l
调控trpedcba的表达;所述启动子pr调控温敏阻遏蛋白
ci
ts
857的表达;所述spp型启动子调控操纵区trpo的基因表达;所述操纵区trpo的下游具有daca基因;所述启动子p
l
和启动子pr的转录方向相反。
9.在一种实施方式中,启动子p
l
的核苷酸序列如seq id no.4所示;启动子pr的核苷酸序列如seq id no.13所示;编码所述启动子spp的核苷酸序列如seq id no.7~11任一所示;操纵区trpo的核苷酸序列如seq id no.3所示;编码所述温敏阻遏蛋白ci
ts
857的核苷酸序列如seq id no.5所示,所述d,d-羧肽酶基因daca的核苷酸序列如seq id no.14所示。
10.在一种实施方式中,所述daca基因的上游还含有trpl碱基序列;所述trpl碱基序列如seq id no.2所示。
11.本发明还一种可调控重组蛋白在大肠杆菌胞外分泌水平的重组大肠杆菌,所述重组大肠杆菌含有所述色氨酸操纵子和温敏阻遏系统共表达的重组质粒,并在基因组上敲除了色氨酸合成酶基因簇trpedcba。
12.在一种实施方式中,所述大肠杆菌可选自:e.coli bl21、e.colijm109、e.coli dh5α、e.coli rosetta、e.coli top10、e.colim110、e.colis110中的任一种。
13.本发明还提供所述重组大肠杆菌的构建方法,所述方法包括构建所述共表达的重组质粒,敲除大肠杆菌基因组的基因簇trpedcba。
14.在一种实施方式中,基因簇trpedcba的敲除方法包括以下步骤:
15.(1)设计并合成含有卡那抗性基因及色氨酸合成酶基因簇trpedcba上下游同源臂的敲除框:上游同源臂-卡纳抗性基因-下游同源臂;
16.(2)将步骤(1)构建的敲除框电转化至含有质粒载体的大肠杆菌电转化感受态细胞,同源重组后筛选出基因整合的大肠杆菌,并消除卡那抗性(kan)基因。
17.本发明还提供一种生产目的蛋白的方法,所述方法是将目的基因在所述重组大肠杆菌中表达。
18.在一种实施方式中,目的蛋白包括但不限于淀粉酶(amyk)、绿色荧光蛋(gfp)、胶原蛋白、果糖基转移酶、甲基对硫磷水解酶、过氧化氢酶、葡萄糖氧化酶等。
19.在一种实施方式中,所述目的蛋白的编码基因连接在质粒上或整合在重组大肠杆菌的基因组上。
20.本发明还提供了一种调控大肠杆菌重组蛋白分泌的方法,所述方法是将所述大肠杆菌在如下条件下发酵:前12h将大肠杆菌在37℃,200bpm振荡培养,抑制daca蛋白表达,促进重组蛋白在胞内积累;12h后控制温度为30℃,200bpm振荡培养,促进daca蛋白表达,并促进胞内重组蛋白的胞外分泌。
21.本发明还提供所述重组质粒或所述重组大肠杆菌在食品、生物领域中的应用。
22.在一种实施方式中,所述应用包括但不限于发酵生产目的蛋白。
23.在一种实施方式中,所述目的蛋白包括但不限于淀粉酶(amyk)、绿色荧光蛋(gfp)、胶原蛋白、果糖基转移酶、甲基对硫磷水解酶、过氧化氢酶、葡萄糖氧化酶等。
24.有益效果:
25.(1)本发明中构建的色氨酸操纵子结合温敏阻遏系统(tat)共表达系统可实现大肠杆菌生长前期能够稳定、正常生长,稳定期促进蛋白的表达;该系统还通过控制发酵温度,调节结构基因的表达水平,调控d,d-羧肽酶实时表达量,增加了胞内可溶性肽聚糖,提高了重组大肠杆菌外膜的渗透性,更加有效的促进重组蛋白的胞外分泌;
ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-egfp。
42.e.coli m9:在e.coli g2基础上含有质粒prsfduet-1
‑△
laci-p
69-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-egfp。
43.e.coli m10:在e.coli g2的基础上含有质粒prsfduet-1
‑△
laci-p
84-trpo-daca-ci
ts
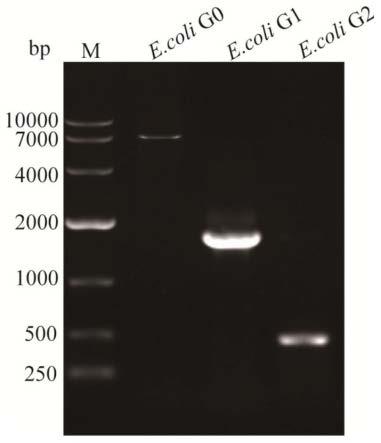
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-egfp。
44.e.coli m11:在e.coli g2的基础上含有质粒prsfduet-1
‑△
laci-p
25-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-amyk。
45.e.coli m12:在e.coli g2的基础上含有质粒prsfduet-1
‑△
laci-p
41-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-amyk。
46.e.coli m13:在e.coli g2的基础上含有质粒prsfduet-1
‑△
laci-p
53-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-amyk。
47.e.coli m14:在e.coli g2的基础上含有质粒prsfduet-1
‑△
laci-p
69-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-amyk。
48.e.coli m15:在e.coli g2的基础上含有质粒prsfduet-1
‑△
laci-p
84-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和petduet-1-pl
‑△
laci-amyk。
49.e.coli g20:e.coli bl21(de3)含有质粒prsfduet-1
‑△
laci-p
53-trpo-egfp-p
t7-trpedcba。
50.e.coli g21:e.coli g2含有质粒prsfduet-1
‑△
laci-p
53-trpo-egfp-p
t7-trpedcba。
51.e.coli g22:e.coli g2含有质粒prsfduet-1
‑△
laci-p
53-trpo-egfp-ci
ts
857-p
r-p
l-trpedcba。
52.e.coli g23:e.coli bl21(de3)含有质粒prsfduet-1
‑△
laci-p
53-trpo-egfp-ci
ts
857-p
r-p
l-trpedcba。
53.2)培养基
54.lb固体培养基:15g/l琼脂,10g/l胰蛋白胨,5g/l酵母提取粉,10g/l nacl,ph 7.0。
55.lb液体培养基:10g/l胰蛋白胨,5g/l酵母提取粉,10g/l nacl,ph 7.0。
56.tb液体培养基:12g/l胰蛋白胨,24g酵母提取粉,甘油4ml,2.31g kh2po4,12.54g k2hpo4,ph 7.5,定容到1l。
57.5хm9(200ml):na2po4·
7h2o 12.8g,kh2po
4 3.0g,nac1 0.5g,nh4cl 1.0g。
58.发酵培养基(1l):200ml 5x m9培养基、2ml mgso4溶液(1mol
·
l-1
)、0.1mlcac2浴液(1mol
·
l-1
)、20ml的20%葡萄糖溶液、1ml硫胺素溶液(1g
·
l-1
)、1ml生物素溶液(1g
·
l-1
)。
59.3)培养方法
60.种子培养:将平板划线长出的单菌落接到lb液体培养基中,培养转速200r/min,在37℃下恒温摇床培养8h。
61.发酵培养:将重组e.coli在lb培养基中37℃培养8h,然后按1%(v/v)的接种量接种到tb培养基中,37℃培养至od
600
=0.8转至25℃诱导表达。氨苄青霉素和氯霉素工作浓度均为50μg
·
ml-1
,卡那霉素工作浓度为25μg
·
ml-1
。
62.4)分析方法
63.生物量测定:测定600nm下的吸光光度值。
64.淀粉酶活性测定:淀粉酶的一个酶活力单位(u)定义:在ph 9.5和50℃下,每分钟水解淀粉释放1μmol还原糖(葡萄糖)所需的酶量。使用改良的二硝基水杨酸(dns)法测定可溶性淀粉水解过程中产生的还原糖。
65.绿色荧光蛋白荧光测定:将菌液在4℃、1
×
104×
g条件下离心10min,将上清与细胞分离,将菌体细胞用10mmol
·
l-1
ph 7.4pbs磷酸盐缓冲液重悬,测定其菌浓od
600 nm
;使用酶标仪和96孔板测定上清中gfp的荧光强度。10mmol
·
l-1
ph 7.4pbs的荧光值作为空白对照。激发和发射波长分别为488nm和533nm。
66.本发明以下实施例中,编码trpedcba序列的核苷酸序列如seq id no.1所示。
67.实施例1重组大肠杆菌中色氨酸合成酶基因簇trpedcba的敲除
68.(1)以质粒pkd13为模板,设计引物并pcr扩增含有kan抗性基因的用于替换trpedcab序列的同源片段,其中kan抗性基因两侧含有frt位点,得到trpedcba序列整合框片段。
69.(2)将pkd46质粒转化至e.coli bl21(de3)感受态细胞,获得e.coli g0菌株,制备电转化感受态细胞,将步骤(1)制得的整合框片段电转化进入e.coli g0感受态中,转化液经过后培养后涂布含有卡那霉素(kan)、氨苄青霉素的lb固体培养基中培养,获得转化子bl21::kan,利用引物trpa-pkd13-fw和trpe-pkd13-rw通过菌落pcr扩增验证trpedcba序列是否成功编辑,筛选出含有卡那霉素抗性基因的阳性转化子e.coli g1。理论上,bl21::kan含有1680bp的同源片段,如图1所示,菌落pcr获得的片段大小与理论上同源片段的大小相符。
70.trpa-pkd13-fw:tgccgccagcggaactggcggctgtgggattaactgcgcgtcgccgctttgtgtaggctggagctgcttc;
71.trpe-pkd13-rw:cccgcctaatgagcgggcttttttttgaacaaaattagagaataacaatgattccggggatccgtcgacc。
72.(3)将步骤(2)构建的e.coli g1接种到无抗性lb培养基培养,取菌液在无抗性lb平板上划线,将单菌落分别对应点在卡那霉素抗性平板和氨苄青霉素抗性平板上,在卡那霉素抗性平板上生长而氨苄青霉素抗性平板上不生长的菌株即为e.coli g1制备e.coli g1转感受态细胞,转化pcp20质粒,涂布于氨苄青霉素抗性、氯霉素抗性双抗性lb培养基平板上,获得含pcp20质粒的阳性转化子,将其接种到无抗性lb培养基中培养,消除pcp20质粒,获得e.coli g2重组大肠杆菌。其中,抗性基因消除后e.coli g2菌株中的同源片段理论上长483bp,菌落pcr扩增验证结果与理论值一致。e.coli g2菌株在kan、氨苄青霉素(amp)平板上均不生长,在无抗性lb平板上正常生长。
73.实施例2稳定期启动子的筛选与表征
74.合成了一个含有40个启动子的文库(部分启动子列表如1所示),用于筛选具有梯度表达水平的不同目标启动子。首先将质粒prsfduet-1上的lac操纵子对应的laci基因片段删除,获得质粒prsfduet-1
‑△
laci。用启动子文库中的40个启动子,分别替换质粒prsfduet-1
‑△
laci中原有的t7启动子,构建获得含有40个不同启动子的质粒文库。以绿色荧光蛋白(egfp)作为模式蛋白,筛选具有梯度表达水平的不同启动子。筛选获得5个不同
egfp表达强度(如图2),且蛋白表达水平集中在稳定期的启动子(p
25
、p
41
、p
53
、p
69
、p
84
),用于后续研究中新型调控系统的构建。
75.表1启动子及序列
76.启动子核苷酸序列p
25
tcttgtcaaattcttaatttggtgctatactggatcgp
41
tcttgtcaaatttttaatgttgtgctatactgtatcgp
53
tctcggcagataccatattatcggctatactgtatcgp
69
tcttgccaaatttgcaaatttgttctatactgtattgp
84
ttttgccagattccctgtgatctgctatactttaaag
77.实施例3新色氨酸操纵子系统设计与应用
78.新色氨酸操纵子系统设计:新色氨酸操纵子系统由敲除了色氨酸合成酶基因簇trpedcba的大肠杆菌细胞和重组质粒组成;所述重组质粒含有t7启动子、spp型启动子、色氨酸合成酶基因簇trpedcba、操纵区trpo和d,d-羧肽酶基因daca;所述操纵区trpo具有可与由阻遏蛋白trpr和色氨酸形成的共价二聚体结合的结合位点;所述t7启动子调控trpedcba的表达;所述spp调控操纵区trpo的基因表达;所述操纵区trpo的下游连接daca基因;所述t7启动子和启动子spp的转录方向相反;所述t7启动子的核苷酸序列如seq id no.6所示;所述spp型启动子的核苷酸序列如seq id no.7~11所示;操纵区trpo如seq id no.3所示;所述daca基因的核苷酸序列如seq id no.14所示。
79.新色氨酸操纵子系统的工作原理为:大肠杆菌基因组含有阻遏蛋白trpr的编码基因(seq id no.12所示),阻遏蛋白trpr能够与高浓度的色氨酸结合形成活性同源二聚体,活性同源二聚可以与trp操纵子的特异性结合位点trpo紧密结合,阻止rna聚合酶的转录。在色氨酸浓度较低时,trpr以一种非活性形式存在,不能与trpo位点结合,此时trp操纵子被rna聚合酶转录。
80.以应用新trp操纵子系统调控daca的表达水平为例(如图3所示),在实施例1的工程菌株e.coli g2中构建新trp操纵子系统,以减少e.coli自身合成的色氨酸对重新构建的trp操纵子的影响。该trp操纵子系统的质粒prsfduet-1
‑△
laci-p
53-trpo-daca-p
t7-trpedcba含有t7启动子、色氨酸合成酶基因簇trpedcba、稳定期启动子p
53
、操纵区trpo和daca基因。t7启动子用于色氨酸合成酶基因簇trpedcba的转录、稳定期启动子用于目的蛋白基因的转录、操纵区trpo用于目的基因转录的调控。在t7启动子的调控下色氨酸合成酶转录表达进而与基因组的抑制物trpr结合形成共价二聚体,特异结合在操纵区trpo上,阻遏由稳定期启动子调控的daca的表达。
81.实施例4色氨酸操纵子抑制系统的可行性验证
82.位于大肠杆菌基因组上的阻遏蛋白(trpr)可与实施例3构建的prsfduet-1
‑△
laci-p
53-trpo-daca-p
t7-trpedcba表达的色氨酸结合,形成活性阻遏物,活性阻遏物可与操纵区trpo特异性结合,形成抑制作用。为了进一步验证通过游离质粒构建色氨酸操纵子阻遏系统是否具有阻遏作用,本实施例选择e.coli g20,e.coli g21,e.coli g213g/l(trp),e.coli g21 5g/l(trp)菌株进行可行性实验。将构建的e.coli g20,e.coli g21分别在m9培养基中37℃发酵培养24h,并针对e.coli g21添加3g/l或5g/l色氨酸以探究外源添加的色氨酸能否和trpr阻遏蛋白结合达到抑制效果。
83.如图3d,基因编辑后不同色氨酸浓度对色氨酸操纵子抑制系统的验证,e.coli g20的荧光值低于e.coli g21的荧光值,说明基因组上能合成色氨酸的初始色氨酸浓度高于e.coli g21。e.coli g21和e.coli g21 3g/l(trp)所展示的荧光值说明外源添加的trp能够和trpr结合形成共价二聚体的阻遏物与色氨酸操纵子特异性结合位点结合,并抑制egfp蛋白的表达。e.coli g21 3g/l(trp)和e.coli g21 5g/l(trp)所抑制egfp的能力相同。上述结果进一步证明:新型trp操纵子系统能够通过色氨酸浓度来抑制egfp的表达,并且具有明显阻遏作用。
84.实施例5色氨酸操纵子抑制系统和温敏阻遏系统的共表达系统设计
85.在实施例3的基础上加入温敏阻遏系统,构建色氨酸操纵子和温敏阻遏系统:色氨酸操纵子和温敏阻遏系统由敲除了色氨酸合成酶基因簇trpedcba的大肠杆菌细胞和重组质粒组成;所述重组质粒含有spp型启动子、低温诱导型启动子pr、高温诱导型启动子p
l
、色氨酸合成酶基因簇trpedcba、操纵区trpo、d,d-羧肽酶基因daca和温敏阻遏蛋白ci
ts
857;所述操纵区trpo具有可与由阻遏蛋白trpr和色氨酸形成的共价二聚体结合的结合位点;所述启动子p
l
上具有温敏阻遏蛋白ci
ts
857的结合位点;所述启动子p
l
调控trpedcba的表达;所述启动子pr调控温敏阻遏蛋白ci
ts
857的表达;所述spp型启动子调控操纵区trpo的基因表达;所述操纵区trpo的下游连接daca基因;所述启动子p
l
和启动子pr的转录方向相反;启动子p
l
的核苷酸序列如seq id no.4所示;启动子pr的核苷酸序列如seq id no.13所示;编码所述启动子spp的核苷酸序列如seq id no.7~11任一所示;操纵区trpo的核苷酸序列如seq id no.3所示;编码所述温敏阻遏蛋白ci
ts
857的核苷酸序列如seq id no.5所示,所述d,d-羧肽酶基因daca的核苷酸序列如seq id no.14所示。
86.在一种实施方式中,色氨酸操纵子和温敏阻遏系统共表达质粒的构建方法为:(1)采用引物d-laci-fw和d-laci-rs,以petduet-1为模板构建获得petduet-1
‑△
laci质粒。采用引物q-laci-fw和q-laci-rs,以采用引物prsfduet-1为模板,构建获得prsfduet-1
‑△
laci质粒。采用引物promoter-25-fw和promoter-25-rs等spp型启动子引物,以prsfduet-1
‑△
laci为模板构建获得含有spp型的prsfduet-1
‑△
laci-p
25
质粒。采用引物trpo-fw和trpo-rs,以prsfduet-1
‑△
laci-p
25
为模板,构建获得prsfduet-1
‑△
laci-p
25-trpo质粒。采用引物daca-fw和daca-rs,以e.coli基因组为模板,扩增基因daca,用限制性内切酶位点ndeⅰ和xhoⅰ连接至质粒prsfduet-1
‑△
laci-p
25-trpo,构建获得重组质粒prsfduet-1
‑△
laci-p
25-trpo-daca。采用引物trpl-daca-fw和trpl-daca-rs,以e.coli基因组为模板,扩增基因trpl-daca,用限制性内切酶位点ndeⅰ和xhoⅰ连接至质粒prsfduet-1
‑△
laci-p
25-trpo,构建获得重组质粒prsfduet-1
‑△
laci-p
25-trpo-trpl-daca。采用引物trp-e-fw和trp-a-rs,以e.coli基因组为模板,扩增基因簇trpedcba,用限制性内切酶位点pstⅰ和kpnⅰ分别与质粒prsfduet-1
‑△
laci-p
25-trpo-daca和prsfduet-1
‑△
laci-p
25-trpo-trpl-daca连接,分别构建获得重组质粒prsfduet-1
‑△
laci-p
25-trpo-daca-trpedcba和prsfduet-1
‑△
laci-p
25-trpo-trpl-daca-trpedcba。采用引物ci857-fw和ci857-rs,以ppl451质粒为模板,扩增基因ci
ts
857,p
l
、pr启动子,利用一步连接方式分别连接至prsfduet-1
‑△
laci-p
25-trpo-daca-trpedcba和prsfduet-1
‑△
laci-p
25-trpo-trpl-daca-trpedcba,分别构建获得重组质粒prsfduet-1
‑△
laci-p
25-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和prsfduet-1
‑△
laci-p
25-trpo-trpl-daca-ci
ts
857-p
r-p
l-trpedcba。
87.按照上述相同的策略构建含稳定期启动子p
41
的质粒prsfduet-1
‑△
laci-p
41-trpo-trpl-daca-ci
ts
857-p
r-p
l-trpedcba、含稳定期启动子p
53
的质粒prsfduet-1
‑△
laci-p
53-trpo-trpl-daca-ci
ts
857-p
r-p
l-trpedcba、含稳定期启动子p
69
的质粒prsfduet-1
‑△
laci-p
69-trpo-trpl-daca-ci
ts
857-p
r-p
l-trpedcba、含稳定期启动子p
84
的质粒prsfduet-1
‑△
laci-p
84-trpo-trpl-daca-ci
ts
857-p
r-p
l-trpedcba。
88.(2)将步骤(1)构建的质粒载体转化至敲除了色氨酸合成酶基因簇trpedcba的大肠杆菌感受态细胞,转化后筛选出含有抗性基因的阳性转化子。
89.trpo-fw:cgaactagttaactagtacgcccatcttagtatattagtta;
90.trpo-rw:gcgtactagttaactagttcgcctatagtgagtcgtattaa;
91.daca-fw:ggaattccatatgatgaataccattttttccgctcg;
92.daca-rw:cggggtaccttaaccaaaccagtgatg;
93.trpl-daca-fw:ggaattccatatgatgaaagcaattttcgtactgaaaggttggtggcgcacttcctgaatgaataccattttttccgctc;
94.trpl-daca-rw:cggggtaccttaaccaaaccagtgatggaaca;
95.trp-e-fw:aactgcagagagaataacaatgcaaacacaaaaaccgactctc;
96.trp-a-rw:cggggtacccggggtaagcgaaacggtaaaaagataaatattaaatga;
97.ci857-fw:ggtcgagatcccggtgcctagtttattgagcgcttatctt;
98.ci857-rw:gtgatacgaaacgaagcatttttgtttaactttaagaaggagaggaattc。
99.promoter-25-fw:aattcttaatttggtgctatactggatcgccccatcttagtata
100.promoter-25-rs:atagcaccaaattaagaatttgacaagaatttcctaatgcaggpromoter-41-fw:aatttttaatgttgtgctatactgtatcgccccatcttagtata
101.promoter-41-rs:atagcacaacattaaaaatttgacaagaatttcctaatgcagg
102.q-lai-fw:cttacattaattgcgttgcgcgggatctcgacgctctcc
103.q-laci-rs:ggagagcgtcgagatcccgcgcaacgcaattaatgtaag
104.d-laci-fw:atacgactcactataggcctctagaaataattttgtttaact
105.d-laci-rs:aattatttctagaggcctatagtgagtcgtattaatttcg
106.温敏阻遏系统的工作原理:该系统可以“打开”或“关闭”重新构建的trp操纵子,用来动态调节daca的表达水平。温敏阻遏蛋白ci
ts
857由低温诱导型启动子pr所调控,当温度不超过30℃时温敏阻遏蛋白ci
ts
857表达并与位于p
l
启动子上的温敏阻遏蛋白结合位点结合,抑制色氨酸基因簇trpedcba的表达。当温度达到42℃时,结合在p
l
启动子上的温敏阻遏蛋白会自行脱落不再阻遏p
l
的转录水平。
107.e.coli的最适生长温度为37℃,温敏抑制系统需要将培养温度设定为42℃,以增加调控色氨酸合成基因簇trpedcba的表达水平。将重组菌株e.coli g22在tb培养基中培养,前12h在42℃,200rpm振荡培养,之后调整温度30℃,200rpm振荡培养。在新型的色氨酸操纵子系统中加入温敏阻遏系统元件,能够达到早期实现细菌的快速生长,后期可以在低温稳定的表达环境中表达daca。同时ci
ts
857的表达可以通过温度诱导来调节,色氨酸的合成可以通过色氨酸操纵子间接调控,从而实现目标蛋白的表达。本实施例还测定了前12h培养温度分别为37℃和42℃对e.coli bl21(de3)菌株细胞生长的影响。结果显示,即使在42℃条件下,e.coli生长也不会受到严重的抑制作用,该系统能够在e.coli中稳态表达。相比
较30℃的条件在42℃下能促进色氨酸合成基因簇的积累并且与trpr结合成共价二聚体,特异结合在trpo阻遏egfp转录。外源添加的0.5g/l trp能够为细菌生长前期提供充足的色氨酸浓度,抑制了目标蛋白egfp的表达。通过调控细菌生长环境的温度实现目的蛋白的高效表达,相比较用iptg试剂表达蛋白的方式更绿色、更安全。
108.通过在不同温度条件下发酵培养(30℃,37℃和42℃),来测试色氨酸操纵子与温敏抑制系统的结合是否符合理论预测且达到抑制效果。从3种不同温度下的研究结果显示,高温42℃确实符合tat系统原理,促进了trpedcba的表达,抑制egfp的转录(48h时,每od
600
下的荧光值为1946.1a.u.),37℃下的抑制效果十分接近42℃的情况(48h时,每od
600
下的荧光值为1475.3a.u.)。相较于30℃条件下,48h时每od
600
下的荧光值为7908.1a.u.)。本实施例还探究了e.coli亲本合成的内源色氨酸对系统的调节能力。结果显示,当系统环境处于e.coli最适生长温度37℃时,e.coli g23自身合成的色氨酸对系统的调节能力相比较e.coli g22更可观,而当系统环境处于42℃时,两者的荧光值相差无异。并且亲本合成的内源性色氨酸不受系统调控,在细菌生长后期会导致系统的色氨酸一直处于高浓度情况,使细菌生长后期无法高效表达目的蛋白。
109.实施例6 tat系统对daca表达的影响
110.在daca的n端加入trpl碱基序列(如seq id no.2所示),具体步骤为:采用引物trpo-fw和trpo-rw,以prsfduet-1为模板,构建获得prsfduet-1-trpo质粒。采用引物trpl-daca-fw和trpl-daca-rw,以e.coli基因组为模板,扩增基因trpl-daca,用限制性内切酶位点ndeⅰ和xhoⅰ连接至质粒prsfduet-1-trpo,构建获得重组质粒prsfduet-1-trpo-trpl-daca。
111.trpo-fw:cgaactagttaactagtacgcccatcttagtatattagtta;
112.trpo-rw:gcgtactagttaactagttcgcctatagtgagtcgtattaa;
113.trpl-daca-fw:ggaattccatatgatgaaagcaattttcgtactgaaaggttggtggcgcacttcctgaatgaataccattttttccgctc;
114.trpl-daca-rw:cggggtaccttaaccaaaccagtgatggaaca;
115.将重组质粒转化至jm109,获得重组菌e.coli g6和e.coli g11。将重组菌在37℃恒温培养箱培养12h,结果显示无论在高拷贝质粒prsfduet-1或petduet-1上表达daca,如图5c,e.coli g6在不添加色氨酸弱化子的情况下,都会导致细菌无法传代,从而无法获得正确的转化子,而在daca的n端加入trpl碱基序列后,细菌可以正常传代,从而获得正确的转化子。通过在tat中加入trpl元件,解决了细菌生产初期因微生物环境中色氨酸浓度较低,不能与trpr形成活性二聚体的问题,实现自诱导型的tat系统表达daca,为提高重组蛋白胞外分泌研究奠定了基础。
116.实施例7 daca过表达对细胞形态的影响
117.本实施例对含有tat系统的菌株e.coli m0、e.coli m7分别在37℃恒温培养箱培养12h而后转30℃恒温培养箱培养12h,观察细胞形态。结果显示(图6a、6c)在37℃下菌株e.coli m0和e.coli m7形态无明显差异。如图6b和图6d,在30℃下,菌株e.coli m0和e.coli m7形态有明显差异。
118.实施例8应用温敏阻遏系统与trp操纵子系统调控目的蛋白的表达
119.以增强型绿色荧光蛋白(enhancing green fluorescent protein,简称egfp),和
淀粉酶amyk为目的蛋白,验证温敏阻遏系统与trp操纵子系统对目的蛋白分泌的调控能力。
120.在e.coli g2的基础上,转入质粒prsfduet-1
‑△
laci-p
25-trpo-daca-ci
ts
857-p
r-p
l-trpedcba和经改造后表达egfp的质粒petduet-1-p
l
‑△
laci-egfp,获得菌株e.coli m6~e.coli m10。
121.将菌株e.coli m6~e.coli m10分别在0~12h于37℃,200rpm振荡培养,12~36h于30℃,200rpm振荡培养,来检测daca对该蛋白胞外分泌能力的影响。如图7,其中菌株e.coli m7的每od
600
胞外荧光值达到11488(a.u.),胞外的荧光占比达到89.8%。加入tat系统的菌株相比较阴性对照菌株的胞外荧光占比有明显的增长,降低了重组蛋白在胞内的过量积累,消除菌体耐受能力降低的弊端,加强了胞外蛋白的分泌,也增强了菌株健康生长的能力。
122.将如genbank登录号kf751392.1所示的基因连接至共表达系统的trpo操纵区的下游,构建重组菌e.coli m11~m17。将上述重组菌分别在37℃条件下培养12h后转入30℃下培养至36h,检测淀粉酶酶活力结果显示12h后30℃条件下培养到36h e.coli m13的酶活可达813.3u
·
g-1
,胞外淀粉酶酶活力占比82.3%,相比较对照菌株,胞外amyk的积累量有了明显的提升。
123.随着蛋白质质量的增大,蛋白质的胞外分泌占比略显下降,表明虽然daca能促进重组蛋白的胞外分泌,但是daca切割肽聚糖链的能力并不能无限放大。
124.对比例:
125.设计引物并pcr扩增含有kan抗性基因的用于替换目的基因dacd的同源片段,采用实施例1中记载的敲除daca或dacb的方法,敲除大肠杆菌中的基因daca,获得突变株bl21-δdacd。将重组质粒petduet-gfp转化至突变株bl21-δdacd后,突变菌株的胞外gfp荧光值为88.1a.u.
·
l
·
g-1
,而对照菌株bl21-petduet-gfp为87.5a.u.
·
l
·
g-1
。可以看出,d,d-羧肽酶dacd基因敲除对大肠杆菌胞外蛋白生产几乎没有效果。
126.虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1