一种微通道连续流制备氯沙坦的方法与流程
1.本发明属于有机合成技术领域,具体涉及一种微通道连续流制备氯沙坦的方法。
背景技术:
2.氯沙坦钾作为最早上市的治疗心血管疾病药物,具有降压效果好,副作用小等特点,自上市以来一直是作为治疗高血压的一线药物,中间体氯沙坦的合成也受到广泛关注,其化学结构如下:
[0003][0004]
目前,合成氯沙坦的方法主要有以下几种:
[0005]
suri babu madasu等(org process res dev,16(2):2025-2030)以2-氰基-4
’‑
甲基联苯和2-丁基-4氯-5-甲酰基咪唑为原料,经溴化反应、n-烷基化反应、还原反应和四氮唑环加成反应得到;
[0006]
徐进宜等(中国药物化学杂志,8(4);271-276)以2-氰基-4
’‑
甲基联苯为原料,经四氮唑环加成反应、三苯甲基保护、溴化反应、n-烷基化反应、还原反应和去保护得到,该工艺操作繁琐,而且需要保护和脱保护步骤,增加生产成本且降低反应收率;
[0007]
robert d.larsen等(j org chem,59(21):6391-6394)报道了另一种方法,使用2-丁基-4-氯-5-甲酰基咪唑和对溴苄溴为起始原料,经n-烷基化反应、还原反应、suzuki偶联和脱保护反应得到氯沙坦,在偶联反应中需要钯催化,且难回收,工业应用受到限制。
[0008]
文献中报道的制备氯沙坦的方法,要么反应时间长,要么操作繁琐,要么安全隐患大。微通道反应器具有分子间扩散快、比表面积大、传热传质高、操作本质安全等优点,经过不断研究、设计,并反复试验及改进,提供一种适合工业化生产的方法。
技术实现要素:
[0009]
针对上述问题,本发明的目的在于提供一种微通道连续流制备氯沙坦的方法。
[0010]
为达到上述面对,提出以下技术方案:
[0011]
一种微通道连续流制备氯沙坦的方法,包括如下步骤:
[0012]
1)将2-氰基-4
’‑
甲基联苯的乙酸溶液与溴代丁二酰亚胺的乙酸溶液分别泵入微通道反应器中混合反应,无催化剂下紫外光引发,保持一定温度和停留时间反应,反应液淬灭后经旋蒸、重结晶后得到2-氰基-4
’‑
溴甲基联苯纯品;
[0013]
2)将步骤1)制备得到的2-氰基-4
’‑
溴甲基联苯纯品与2-丁基-4-氯-5-甲酰基咪
唑溶于甲苯中,同时配制naoh和tbab的混合水溶液,将上述配制的甲苯溶液和混合水溶液分别泵入微通道反应器进行反应,保持一定温度和停留时间反应,反应完成后进行分离得到淡黄色甲苯相,该有机相不做进一步后处理,加硼氢化钠和甲醇后室温搅拌至反应完全,加适量去离子水析晶后得到2-丁基-4-氯-5-羟甲基-1-(2
′‑
氰基-联苯基-4-)甲基咪唑粗品;
[0014]
3)将步骤2)制备得到的粗品烘干,直接加入有机溶剂配制成溶液;同时配nan3和et3n
·
hcl的混合水溶液,将上述两种溶液分别泵入微通道反应器中进行四氮唑环加成反应,保持一定温度和停留时间反应,反应完成后通过萃取分离得到淡黄色固体,即氯沙坦粗品;
[0015]
4)步骤3)制备得到的氯沙坦粗品经甲醇重结晶后得到氯沙坦纯品。
[0016]
进一步地,紫外光照射所采用的紫外光为200~450nm紫外光,优选为220~375nm。
[0017]
进一步地,步骤1)中的2-氰基-4
’‑
甲基联苯的乙酸溶液的浓度为0.10~0.40mol/l,溴代丁二酰亚胺的乙酸溶液的浓度为0.10~0.50mol/l,2-氰基-4
’‑
甲基联苯和溴代丁二酰亚胺的摩尔比为1.00:1.00~1.25。
[0018]
进一步地,步骤1)中物料在微通道反应器中的反应温度为65~115℃,停留时间为60~180s,优选为80~90℃,105~135s。
[0019]
进一步地,步骤2)中配制的甲苯溶液中的2-氰基-4
’‑
溴甲基联苯纯品与2-丁基-4-氯-5-甲酰基咪唑的摩尔比为1.00:1.00~1.20,优选为1.00:1.00~1.05,2-氰基-4
’‑
溴甲基联苯纯品的浓度为0.50~1.00mol/l;配制的混合水溶液中naoh和tbab的摩尔比为1.00:0.10~0.50,naoh浓度为0.50~2.00mol/l。
[0020]
进一步地,步骤2)中物料在微通道反应器中的反应温度为70~95℃,反应停留时间为90~210s。
[0021]
进一步地,步骤3)中的有机溶剂为甲苯、氯苯、正丁醇或二甲苯,2-丁基-4-氯-5-羟甲基-1-(2
′‑
氰基-联苯基-4-)甲基咪唑粗品的浓度为0.10~0.50mol/l;混合水溶液中的nan3和et3n
·
hcl的摩尔比为1.00:0.50~1.50,nan3的浓度为0.50~4.00mol/l。
[0022]
进一步地,步骤3)中的反应温度为130~160℃,反应停留时间为45-120min,优选为140~150℃,60~90min,背压阀压力为8~12bar。
[0023]
进一步地,步骤3)中2-丁基-4-氯-5-羟甲基-1-(2
′‑
氰基-联苯基-4-)甲基咪唑粗品与nan3的摩尔比为1.00:1.00~2.00。
[0024]
进一步地,步骤2)和步骤3)中的微通道反应器的内径为0.50-5.00mm,连续流微结构反应器可以采用微通道反应器,微尺度或介尺度的管式反应器;微反应器可选市售品牌(进口品牌chemtrix plantrix mr260或mr555;国产金德新材料c系列,以及山东微井、杭州沈氏等类似型号的国产品牌),也可按需设计定制加工;市售微反应器或定制管式反应器的通道直径不超过3/8英寸,带强化设备的管式反应器不超过1英寸,线性长度一般不超过100米;对于定制管式反应器,根据需要可加入静态混合器强化。反应器的材质不限于碳化硅、石墨、石英、玻璃等无机材料,也不限于不锈钢、哈氏合金、钛合金、蒙乃尔合金等金属材料。
[0025]
相对于现有技术,本发明的有益效果在于:
[0026]
1)本发明提供的氯沙坦的连续流制备方法,组合优化的三步连续流微反应技术,能够缩短反应时间,大大提高生产效率;另外,反应液的淬灭和分离也可实现连续化,简化
操作适合工业化生产;
[0027]
2)连续流合成方法具有安全可靠,重现性优,放大效应不明显的特点,在节能减排和绿色环保上具有很大的优势;进一步优化可实现本质安全,黑灯操作的变革;
[0028]
3)产量随泵流通量、微反应器的增大而增大。
具体实施方式
[0029]
下文将结合具体实施例对本发明的制备方法做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0030]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
[0031]
采用本发明的方法制备氯沙坦的合成路线如下所示:
[0032][0033]
实施例1
[0034]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;将两种溶液经注射泵都以3.30μl/min的流速泵入微通道反应器中,2-氰基-4
’‑
甲基联苯与溴代琥珀酰亚胺的摩尔比为1:1.05,反应温度为105℃,反应液停留时间为90s,以365nm的紫外灯引发反应,反应流经冰水淬灭后萃取、蒸馏、重结晶得2-氰基-4
’‑
溴甲基联苯纯品,收率不低于83.5%,归一法纯度不低于99.0%。化合物ⅰ结构表征:1h nmr(400mhz,cdcl3)δ:7.77(dd,j=7.7,1.4hz,1h),7.65(td,j=7.7,1.4hz,1h),7.56-7.50(m,5h),7.46(td,j=7.7,1.3hz,1h),4.55(s,2h).
13
c nmr(101mhz,cdcl3)δ:144.84,138.45,138.31,133.94,133.03,130.14,129.56,129.34,127.93,118.70,111.36,32.94.
[0035]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯纯品与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,摩尔比为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯浓度为1.00mol/l;配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为1.30mol/l,将甲苯溶液和水溶液两种溶液都以8.35ml/min的流速,泵入微反应器中,微通道反应器的内径截面为2mm
×
2mm,反应温度85
±
1℃,反应液停留时间120s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于84.2%,归一法纯度不低于99.0%。化合物ⅱ结构表征:1h nmr(400mhz,cdcl3)δ:7.75(dd,j=7.8,1.3hz,1h),7.64(td,j=7.7,1.4hz,1h),7.51(d,2h),7.48-7.42(m,2h),7.11(d,2h),5.30(s,2h),4.50(s,2h),2.57(t,j=1.4hz,2h),1.65-1.63(m,2h),1.36-1.30(m,2h),0.86(t,j=7.4hz,3h).
13
c nmr(101mhz,cdcl3)δ:148.76,144.72,137.89,136.89,133.91,133.05,130.09,129.51,127.93,127.39,126.38,125.07,118.66,111.29,53.18,47.31,29.81,26.86,22.51,13.83.
[0036]
3)将步骤2)所得固体,溶于正丁醇并配制成0.10mol/l的溶液;同时配制nan3和et3n
·
hcl的水溶液,前后两者摩尔比为1.00:1.00,其中nan3浓度为0.80mol/l。将正丁醇溶
液和水溶液分别以1.51ml/min、0.37ml/min的流速注入微反应器中,微通道反应器的内径截面为2mm
×
2mm,反应温度140℃,反应液停留时间90min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后有机相旋蒸得到氯沙坦粗品,收率不低于68.4%。化合物ⅲ结构表征:1h nmr(400mhz,dmso-d6)δ:16.39(s,1h),7.68-7.64(m,2h),7.59-7.51(m,2h),7.07(d,2h),7.02(d,2h),5.26(s,1h),5.24(s,2h),4.32(s,2h),2.45(t,j=7.5hz,2h),1.46-1.41(m,2h),1.25-1.20(m,2h),0.79(t,j=7.3hz,3h).
13
c nmr(101mhz,dmso-d6)δ:148.76,144.72,137.89,136.89,133.91,133.05,130.09,129.51,127.93,127.39,126.38,125.07,118.66,111.29,53.18,47.31,29.81,26.86,22.51,13.83.
[0037]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,总收率不少于48.0%(以2-氰基-4
’‑
甲基联苯计)。
[0038]
实施例2
[0039]
1)配制0.10mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.10mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯与溴代琥珀酰亚胺的摩尔比为1.00:1.00,将两种溶液经注射泵都以2.50μl/min的流速泵入微通道反应器中,反应温度为100℃,反应液停留时间为120s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于83.3%,纯度不低于99.7%。
[0040]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯纯品与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,前后两者摩尔比为1.00:1.00,其中2-氰基-4
’‑
溴甲基联苯浓度为0.50mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.50,其中氢氧化钠浓度为0.50mol/l,将制备的甲苯溶液和水溶液都以7.76ml/min的流速,泵入微反应器中,微通道反应器的内径截面为2mm
×
2mm,反应温度85℃,反应液停留时间135s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于82.5%,纯度不低于99.3%。
[0041]
3)将步骤2)所得固体,使用正丁醇溶剂溶解配制0.10mol/l的溶液;同时配制nan3和et3n
·
hcl的水溶液,前后两者摩尔比为1.00:1.00,其中nan3浓度为0.50mol/l,将该正丁醇溶液和水溶液分别以1.51ml/min、0.37ml/min的流速注入微反应器中,微通道反应器的内径截面为2mm
×
2mm,反应温度145℃,反应液停留时间90min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液旋蒸得到氯沙坦粗品,收率不低于74.1%。
[0042]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.6%,总收率不少于50.5%(以2-氰基-4
’‑
甲基联苯计)。
[0043]
实施例3:
[0044]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯与溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以1.66μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为180s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于84.8%,纯度不低于99.4%。
[0045]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,前后两者摩尔比为1.00:1.10,其中2-氰基-4
’‑
溴甲基联苯的浓度为1.00mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中氢氧化钠浓度为
1.10mol/l,将甲苯溶液和水溶液都以5.56ml/min的流速,泵入微反应器中,微通道反应器的内径截面为2mm
×
2mm,反应温度95℃,反应液停留时间180s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于90.7%,纯度不低于99.3%。
[0046]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l,将该正丁醇溶液和水溶液分别以1.51ml/min、0.37ml/min的流速注入微反应器中,微通道反应器的内径截面为2mm
×
2mm,反应温度150℃,反应液停留时间90min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后旋蒸得到氯沙坦粗品,收率不低于68.5%。
[0047]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后,冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,总收率不少于50.0%(以2-氰基-4
’‑
甲基联苯计)。
[0048]
对比例1:传统釜式工艺
[0049]
取2-氰基-4
’‑
甲基联苯1.93g(10mmol)、溴代琥珀酰亚胺1.96g(11mmol)溶于50ml乙酸中,常温搅拌下使用365nm紫外光照射两个小时,反应液后处理得到2-氰基-4
’‑
溴甲基联苯纯品2.17g,该纯品与2-丁基-4-氯-5-甲酰基咪唑按1.00:1.05的摩尔比溶于甲苯中,同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh加入的量为10mmol,前面配制的两种溶液混合后置于70℃水浴锅中保温搅拌三个小时,反应结束后分离得到有机相,加入硼氢化钠还原,析晶后得到2-丁基-4-氯-5-羟甲基-1-(2
′‑
氰基-联苯基-4-)甲基咪唑固体,烘干后称量为2.39g,将该固体溶于正丁醇溶剂中;同时向溶剂中加入nan3和et3n
·
hcl,两者摩尔比为1.00:1.00,其中nan3加入15mmol,将反应装置置于油浴锅中120℃回流24小时以上,反应液后处理得1.51g氯沙坦,总收率35.7%(以2-氰基-4
’‑
甲基联苯计)。
[0050]
由实施例1-3与对比例1对比可知,采用本发明的方法配合三步连续流微反应技术相比釜式工艺能够明显提高收率。
[0051]
实验一,微反应器反应温度范围验证实验;
[0052]
实施例4
[0053]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯与溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以3.30μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为90s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于85.7%,纯度不低于98.5%。
[0054]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为1.00mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为1.10mol/l,将这两种溶液都以11.13ml/min的流速,泵入微反应器中,反应温度85℃,反应液停留时间90s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后通过液相色谱测定,流出液收率不少于78.3%,纯度不低于99.3%。
[0055]
3)将步骤2)所得固体,配制成0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl
的混合水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.8mol/l,将正丁醇溶液和水溶液分别以2.26ml/min、0.56ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间60min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后旋蒸得到氯沙坦粗品,收率不低于66.4%。
[0056]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,总收率不少于44.5%(以2-氰基-4
’‑
甲基联苯计)。
[0057]
实施例5:
[0058]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和溴代琥珀酰亚胺的摩尔比为1:1.05,将前面配制的两种溶液经注射泵都以3.30μl/min的流速泵入微通道反应器中,反应温度为100℃,反应液停留时间为90s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于84.7%,纯度不低于99.8%。
[0059]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为1.00mol/l;同时配制naoh和tbab的水溶液,两者摩尔比为1.00:0.10,其中naoh浓度为1.10mol/l。将这两种溶液都以11.13ml/min的流速泵入微反应器中,反应温度90℃,反应液停留时间90s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于77.1%,纯度不低于99.3%。
[0060]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l。将该正丁醇溶液和水溶液分别以2.26ml/min、0.56ml/min的流速注入微反应器中,反应温度150℃,反应液停留时间60min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后旋蒸得到氯沙坦粗品,收率不低于62.2%。
[0061]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于40.7%(以2-氰基-4
’‑
甲基联苯计)。
[0062]
实施例6:
[0063]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以3.30μl/min的流速泵入微通道反应器中,反应温度为90℃,反应液停留时间为90s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于81.0%,纯度不低于99.4%。
[0064]
2)将步骤2)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为1.00mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为1.10mol/l,将这两种溶液都以11.13ml/min的流速,泵入微反应器中,反应温度80℃,反应液停留时间90s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于74.6%,纯度不低于99.3%。
[0065]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l,将该正丁醇溶液和水溶液
分别以2.26ml/min、0.56ml/min的流速注入微反应器中,反应温度140℃,反应液停留时间60min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后旋蒸得到氯沙坦粗品,收率不低于63.3%。
[0066]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后,冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.06%,收率不少于37.8%(以2-氰基-4
’‑
甲基联苯计)。
[0067]
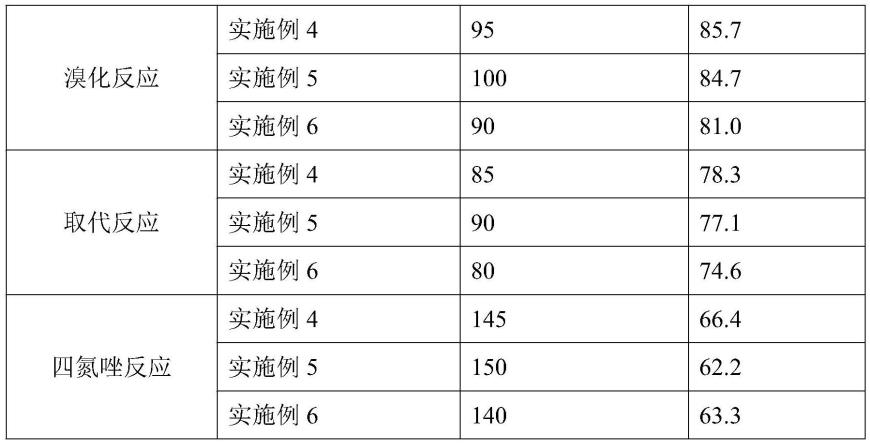
表1不同反应温度对产品收率的影响结果
[0068][0069][0070]
从表1的结果分析:由实施例4-6可知,在物料配比及反应时间一定情况下,在微通道反应器中进行自由基溴化反应的优选温度为95℃,进行n-烷基化反应的优化温度为85℃;进行四氮唑反应的优化温度为145℃,当温度超出优选温度范围后,产物收率会有所下降。
[0071]
对比例2:
[0072]
1)配制0.40mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.50mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和溴代琥珀酰亚胺的摩尔比为1.00:1.25,将两种溶液经注射泵都以3.30μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为90s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体,收率不低于71.38%,纯度不低于99.73%。
[0073]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.00,其中2-氰基-4
’‑
溴甲基联苯的浓度为1.00mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为2.00mol/l,将这两种溶液都以11.13ml/min的流速,泵入微反应器中,反应温度85℃,反应液停留时间90s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于67.1%,纯度不低于99.8%。
[0074]
3)将步骤2)所得固体,配制0.50mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为4.00mol/l,将该正丁醇溶液和水溶液分别以2.26ml/min、0.56ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间
60min,反应压力10bar,反应液流入次氯酸钠水溶液中,分液后旋蒸得到氯沙坦粗品,收率不低于56.1%。
[0075]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于26.9%(以2-氰基-4
’‑
甲基联苯计),采用的微反应器为杭州沈氏定制设备(型号wrc13000;尺寸,直径小于1000μm)
[0076]
实验二:微反应器反应停留时间范围验证实验
[0077]
实施例7:
[0078]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以2.50μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为120s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体,收率不低于93.3%,纯度不低于99.0%。
[0079]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为0.50mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为0.55mol/l,将这两种溶液都以7.41ml/min的流速,泵入微反应器中,反应温度85℃,反应液停留时间135s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于93.6%,纯度不低于99.0%。
[0080]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l,将该正丁醇溶液和水溶液分别以1.81ml/min、0.45ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间75min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后旋蒸得到氯沙坦粗品,收率不低于82.5%。
[0081]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于67.0%(以2-氰基-4
’‑
甲基联苯计)。
[0082]
实施例8:
[0083]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以2.85μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为105s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于85.5%,纯度不低于98.5%。
[0084]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为0.50mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为0.55mol/l,将这两种溶液都以8.35ml/min的流速,泵入微反应器中,反应温度85℃,反应液停留时间120s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于87.3%,纯度不低于99.0%。
[0085]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l,将该正丁醇溶液和水溶液
分别以2.26ml/min、0.56ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间60min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后有机相旋蒸得到氯沙坦粗品,收率不低于66.4%。
[0086]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于49.5%(以2-氰基-4
’‑
甲基联苯计)。
[0087]
实施例9:
[0088]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以2.22μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为135s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于88.2%,纯度不低于99.0%。
[0089]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为0.50mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为0.55mol/l。将这两种溶液都以6.68ml/min的流速泵入微反应器中,反应温度85℃,反应液停留时间150s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于90.5%,纯度不低于99.0%。
[0090]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l,将该正丁醇溶液和水溶液分别以1.51ml/min、0.37ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间90min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后有机相旋蒸得到氯沙坦粗品,收率不低于74.2%。
[0091]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于58.0%(以2-氰基-4
’‑
甲基联苯计)。
[0092]
对比实施例3:
[0093]
1)配制成0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯和溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以3.33μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为90s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于84.6%,纯度不低于99.0%。
[0094]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为0.50mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为0.55mol/l。将这两种溶液都以11.13ml/min的流速泵入微反应器中,反应温度85℃,反应液停留时间90s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于80.0%,纯度不低于99.0%。
[0095]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l。将正丁醇溶液和水溶液分别以3.00ml/min、0.75ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间
45min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后有机相旋蒸得到氯沙坦粗品,收率不低于42.6%。
[0096]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于28.8%(以2-氰基-4
’‑
甲基联苯计)。
[0097]
对比实施例4:
[0098]
1)配制0.20mol/l的2-氰基-4
’‑
甲基联苯的乙酸溶液,0.21mol/l的溴代琥珀酰亚胺的乙酸溶液;2-氰基-4
’‑
甲基联苯与溴代琥珀酰亚胺的摩尔比为1:1.05,将两种溶液经注射泵都以2.00μl/min的流速泵入微通道反应器中,反应温度为95℃,反应液停留时间为150s,提前开启365nm紫外灯照射微反应板,反应液经冰水淬灭后萃取、重结晶得2-氰基-4
’‑
溴甲基联苯固体。收率不低于88.0%,纯度不低于99.0%。
[0099]
2)将步骤1)所得2-氰基-4
’‑
溴甲基联苯固体与2-丁基-4-氯-5-甲酰基咪唑溶于甲苯中,两者摩尔比例为1.00:1.05,其中2-氰基-4
’‑
溴甲基联苯的浓度为0.50mol/l;同时配制naoh和tbab的水溶液,naoh和tbab的摩尔比为1.00:0.10,其中naoh浓度为0.55mol/l,将这两种溶液都以5.56ml/min的流速泵入微反应器中,反应温度85℃,反应液停留时间180s,收集反应液,分离得到有机相,有机相中直接加入硼氢化钠还原,反应完后加蒸馏水析晶后得到白色固体,过滤、干燥后收率不低于89.0%,纯度不低于99.0%。
[0100]
3)将步骤2)所得固体,配制0.10mol/l的正丁醇溶液;同时配制nan3和et3n
·
hcl的水溶液,两者摩尔比例为1.00:1.00,其中nan3浓度为0.80mol/l,将该正丁醇溶液和水溶液分别以1.18ml/min、0.29ml/min的流速注入微反应器中,反应温度145℃,反应液停留时间115min,反应压力10bar,反应液直接流入次氯酸钠水溶液中淬灭,分液后有机相旋蒸得到氯沙坦粗品,收率不低于63.5%。
[0101]
4)氯沙坦粗品加入5倍量的甲醇,加热溶解后冷却析出固体,过滤得氯沙坦纯品,纯度不低于99.0%,收率不少于48.5%(以2-氰基-4
’‑
甲基联苯计)。
[0102]
表2不同停留时间对产品收率的影响结果
[0103]
[0104][0105]
由表2的实验结果分析:由实施例7-9和对比例3-4可知,在物料配比和温度一定情况下,在微通道反应器中进行自由基溴化反应的优选时间为105-135s,进行n-烷基化反应的优化时间为120-150s;进行四氮唑反应的优化时间为60-90min。当时间超出优选停留时间范围后,产物收率会有明显下降。
[0106]
本发明提供了一种通过连续流工艺生产氯沙坦的方法,通过该方法提高了反应选择性,减少了反应能耗,缩短了反应时间,增加反应安全性,提高了生产效率,具有潜在的工业化价值。
[0107]
以上显示和描述了本发明的基本原理、主要特征和优点。本行业的技术人员应该了解,上述实施例不以任何形式限制本发明,凡采用等同替换或等效变换的方式所获得的技术方案,均落在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1