一种复合催化剂、二氧化碳基共聚物及其制备方法和应用与流程
1.本发明属于生物降解材料技术领域,具体涉及一种复合催化剂、二氧化碳基共聚物及其制备方法和应用。
背景技术:
2.随着工业化进程的迅猛发展,化石能源的大量开发利用,不可避免地导致大气中二氧化碳含量大幅上升并带来空气污染等一系列问题。自此温室气体二氧化碳的排放也得到了广泛关注,进而二氧化碳固定和利用成为当前的热点领域,而以二氧化碳为单体合成高分子可生物降解材料已然成为二氧化碳高值利用的重要研究方向。
3.作为二氧化碳共聚物的代表,聚碳酸亚丙酯(ppc)是二氧化碳与环氧丙烷的共聚物,是一种完全可生物降解材料。然而,现有工业化工艺由于采用非均相催化(三元稀土催化剂、戊二酸锌催化剂以及双金属催化剂等)为主,在制备过程中,副产物碳酸丙烯酯(cpc)生成量较大,一般在6%以上,且cpc沸点较高,与ppc相容性好,从ppc中分离cpc的能耗超过聚合工段,从而大幅度增加ppc的生产能耗和成本,另一方面,若不彻底除去ppc中的小分子cpc,其在后续加工和使用过程中的外迁移、水解及挥发性又会带来产品气味、强度及封口性差等一系列问题。因此,ppc选择性较差已经成为困扰ppc产业升级和发展的关键难题。
4.cn112358607a公开了一种溶液法合成高分子量ppc的方法,其中双金属催化剂可实现环氧丙烷的完全转化,同时由于反应过程中溶剂的存在,cpc生成量有所降低,但是其制备方法中副产物cpc的含量依然高于2%,依然需要后续进一步处理。并且,虽然采用均相的卟啉钴或salen钴体系催化剂,可以实现二氧化碳与环氧丙烷的高选择性聚合,聚合过程中cpc生成量低于0.5%,但由于均相催化剂合成比较困难,同时产物颜色难以彻底去除,目前尚未实现工业应用。
5.因此,开发一种可以降低cpc生成量的复合催化剂,是目前该领域工业生产中悬而未决的难题之一。
技术实现要素:
6.针对现有技术的不足,本发明的目的在于提供一种复合催化剂、二氧化碳基共聚物及其制备方法和应用,所述复合催化剂包括非均相催化剂和环状酸酐的组合;所述复合催化剂在用于二氧化碳基共聚物的制备时,可使得聚合链增长的端基保持为无回咬的羧酸金属端基,进而大幅降低了产物中副产物的生成量。
7.为达到此发明目的,本发明采用以下技术方案:
8.第一方面,本发明提供一种复合催化剂,所述复合催化剂包括非均相催化剂和环状酸酐的组合。
9.本发明提供的复合催化剂包括非均相催化剂和环状酸酐的组合,所述复合催化剂可以用于二氧化碳基共聚物的制备,且所述复合催化剂用于制备二氧化碳基共聚物时,可使得聚合物链增长的端基保持为无回咬的羧酸金属端基,取代了常规催化剂催化环氧化物
和二氧化碳共聚形成的易回咬羧基金属端基,进而大幅降低了产物中副产物(环状碳酸酯)的含量,提高了环氧化物的转化率以及二氧化碳基共聚物的选择性。
10.优选地,所述非均相催化剂包括二元羧酸锌催化剂、稀土三元催化剂或双金属催化剂中的任意一种或至少两种的组合。
11.本发明对非均相催化剂的具体来源并不作限定,市售或依照现有技术中提供的方法制备得到的非均相催化剂均可。其中,优选按照专利cn1250603c提供的制备方法合成得到的稀土三元催化剂,优选按照专利cn105418907a中实施例1的制备方法制备得到的二元羧酸锌催化剂,优选按照专利cn102432857a实施例1的制备方法制备得到的双金属催化剂。
12.在本发明中对于所提供的复合催化剂的制备方法也不做特殊的限定,可以将非均相催化剂与环状酸酐通过水浴震荡、研磨或球磨的方法进行复配。
13.优选地,所述环状酸酐包括马来酸酐、丁二酸酐、戊二酸酐、3-乙基-3-甲基戊二酸酐、环丁烷-1,2-二甲酸酐、衣康酸酐、甲基丁二酸酐、烯丙基丁二酸酐、环戊烷-1,2-二甲酸酐、4-甲基-1,2-环己二羧酸酐、降冰片烯二酸酐、甲基-5-降冰片烯-2,3-二羧酸酐、邻苯二甲酸酐、1,2,3,6-四氢苯酐、3,4,5,6-四氢苯酐、甲基四氢邻苯二甲酸酐、1,2-环己二甲酸酐、十二烷基琥珀酸酐或2,2'-联苯二甲酸酐中的任意一种或至少两种的组合。
14.第二方面,本发明提供一种二氧化碳基共聚物,所述二氧化碳基共聚物的制备原料包括如第一方面所述的复合催化剂、二氧化碳和环氧化物的组合。
15.本发明提供的二氧化碳基共聚物的制备原料包括如第一方面所述的复合催化剂、二氧化碳和环氧化物的组合;其中,所述二氧化碳和环氧化物在所述复配催化剂中的非均相催化剂以及环状酸酐的复合催化作用下进行开环(共)聚合,进而即可制备得到所述二氧化碳基共聚物;利用所述复合催化剂中包含的环状酸酐,从而在产物链增长的端基可以保持为无回咬的羧酸金属端基,取代环氧化物和二氧化碳共聚形成的易回咬羧基金属端基,进而大幅降低产物中副产物(环状碳酸酯)的生成量。
16.优选地,所述二氧化碳基共聚物的链段中碳酸酯单元的摩尔百分含量高于90%,例如91%、92%、93%、94%、95%、96%、97%、98%或99%等。
17.优选地,所述环氧化物包括环氧乙烷、1,2-环氧丙烷或1,2-环氧丁烷中的任意一种或至少两种的组合。
18.优选地,所述复合催化剂中的环状酸酐与环氧化物的摩尔比为(0.1~4):100,例如0.3:100、0.6:100、0.9:100、1:100、1.2:100、1.5:100、1.8:100、2:100、2.1:100、2.4:100、2.7:100、3.1:100、3.4:100、3.7:100或3.9:100等,进一步优选为(0.5~3.5):100。
19.优选地,所述二氧化碳基共聚物的制备原料中还包括溶剂。
20.优选地,所述溶剂包括碳酸二甲酯、碳酸二乙酯、丙酮、丁酮、二甲基甲酰胺、二氯甲烷、三氯甲烷、四氢呋喃、1,3-二氧五环或二氧六环中的任意一种或至少两种的组合。
21.优选地,所述环氧化物与溶剂的体积比为1:(0.5~4),例如1:1、1:1.5、1:2、1:2.5、1:3或1:3.5等,进一步优选为1:(0.8~3),更进一步优选为1:(1~2)。
22.优选地,所述环氧化物和溶剂中的水含量均低于50ppm,例如45ppm、40ppm、35ppm、30ppm、28ppm、26ppm、24ppm、22ppm、20ppm、18ppm、16ppm、14ppm或12ppm等。
23.第三方面,本发明提供一种如第二方面所述二氧化碳基共聚物的制备方法,所述制备方法包括:将复合催化剂、环氧化物和二氧化碳在溶剂中进行聚合反应,得到所述二氧
化碳基共聚物。
24.在本发明中,对于物料的加入顺序不做限定,优选为依次加入溶剂、环氧单体、复合催化剂、二氧化碳这一顺序;并且制备过程优选在高压反应釜中进行,但是本发明对所述高压反应釜的具体型号与规格不作限定,优选为配有机械搅拌及内冷盘管的高压反应釜。
25.优选地,所述聚合反应的产物中环状碳酸酯的摩尔百分含量低于1.5%(以环状碳酸酯和聚合物中所有重复单元的总摩尔数计算),所述二氧化碳基共聚物的数均分子量为200000~600000g/mol,例如220000、240000、260000、280000、300000、320000、340000、360000、380000、400000、420000或440000等。
26.优选地,所述二氧化碳基共聚物的分子量分布为1.1~4.5,例如2.2、2.4、2.6、2.8、3、3.2、3.4、3.6、3.8、4、4.2或4.4等。
27.优选地,所述复合催化剂中的非均相催化剂为二元羧酸锌催化剂,所述复合催化剂在聚合反应的初始浓度为2~80mg/ml,例如4mg/ml、6mg/ml、8mg/ml、10mg/ml、12mg/ml、14mg/ml、16mg/ml、18mg/ml、20mg/ml、25mg/ml、30mg/ml、35mg/ml、40mg/ml、45mg/ml、50mg/ml、60mg/ml或70mg/ml等,进一步优选为6~70mg/ml,更进一步优选为10~60mg/ml。
28.优选地,所述复合催化剂中的非均相催化剂为稀土三元催化剂,所述复合催化剂在聚合反应的初始浓度为4~120mg/ml,例如5mg/ml、7mg/ml、10mg/ml、13mg/ml、16mg/ml、18mg/ml、21mg/ml、23mg/ml、26mg/ml、29mg/ml、31mg/ml、35mg/ml、40mg/ml、50mg/ml、60mg/ml、70mg/ml、80mg/ml、90mg/ml、100mg/ml或110mg/ml等,进一步优选为8~100mg/ml,更进一步优选为10~80mg/ml。
29.优选地,所述复合催化剂中的非均相催化剂为双金属催化剂,所述复合催化剂在聚合反应的初始浓度为0.05~120mg/ml,例如0.4mg/ml、0.6mg/ml、0.8mg/ml、1mg/ml、1.2mg/ml、1.4mg/ml、1.6mg/ml、1.8mg/ml、1mg/ml、10mg/ml、30mg/ml、50mg/ml、70mg/ml、90mg/ml或110mg/ml等,进一步优选为0.1~100mg/ml,更进一步优选为0.2~95mg/ml。
30.优选地,所述复合催化剂中的非均相催化剂为稀土三元催化剂或二元羧酸锌催化剂,所述聚合反应的温度为70~100℃,例如73℃、76℃、79℃、82℃、85℃、88℃、91℃、94℃或97℃等,进一步优选为75~95℃,更进一步优选为80~90℃。
31.优选地,所述复合催化剂中的非均相催化剂为稀土三元催化剂或二元羧酸锌催化剂,所述聚合反应的压力为4~10mpa,例如4.5mpa、5mpa、5.5mpa、6mpa、6.5mpa、7mpa、7.5mpa、8mpa、8.5mpa、9mpa或9.5mpa等,进一步优选为6~9mpa,更进一步优选为7.5~8.5mpa。
32.优选地,所述复合催化剂中的非均相催化剂为稀土三元催化剂或二元羧酸锌催化剂,所述聚合反应的时间为12~36h,例如14h、16h、18h、20h、22h、24h、26h、28h、30h、32h或34h等,进一步优选为14~30h,更进一步优选为16~24h。
33.优选地,所述复合催化剂中的非均相催化剂为双金属催化剂,所述聚合反应的温度为40~70℃,例如42℃、44℃、46℃、48℃、50℃、52℃、54℃、56℃、58℃、60℃或65℃等,进一步优选为50~68℃,更进一步优选为53~67℃。
34.优选地,所述复合催化剂中的非均相催化剂为双金属催化剂,所述聚合反应的压力为2~10mpa,例如3mpa、3.5mpa、4.5mpa、5mpa、5.5mpa、6mpa、6.5mpa、7mpa、7.5mpa、8mpa、8.5mpa、9mpa或9.5mpa等,进一步优选为3~9mpa,更进一步优选为4~8mpa。
35.优选地,所述复合催化剂中的非均相催化剂为双金属催化剂,所述聚合反应的时间为4~24h,例如10h、12h、14h、16h、18h、20h或22h等,进一步优选为5~20h,更进一步优选为6~18h。
36.优选地,所述聚合反应后还包括冷却和脱挥的步骤。
37.在本发明中,所述冷却即聚合反应结束后,将高压反应釜的釜外夹套循环水由热水切换成冷水,并在反应釜内冷盘管中通入冷却液,保持釜内反应产物搅拌状态下在30min内迅速冷却至40℃以下;所述脱挥即共聚产物自反应釜内取出后置于真空烘箱内40℃,-0.1mpa条件下脱除溶剂,得到共聚产物。
38.优选地,所述制备方法具体包括如下步骤:
39.(1)将复合催化剂、部分溶剂以及部分环氧化物混合后,通入二氧化碳进行预聚反应,得到预聚物;
40.(2)将剩余溶剂和剩余环氧化物加入步骤(1)得到的预聚物中,通入二氧化碳进行反应,得到所述二氧化碳基共聚物。
41.作为本发明的优选技术方案,本发明提供的二氧化碳基共聚物的制备方法采用溶液聚合工艺,在一定的复合催化剂浓度下引发聚合反应后,将环氧化物、溶剂以一定比例混合后连续或分步投入,再通入二氧化碳来维持反应压力的恒定。
42.优选地,在本发明提供的优选制备方法的步骤(1)中部分环氧化物投入量占总环氧化物质量百分含量为的25~50%,例如28%、31%、34%、37%、40%、43%、46%或49%等,进一步优选为28~40%,更进一步优选为30%~35%;相应的,步骤(1)中部分溶剂与环氧化物等体积投入。
43.优选地,步骤(2)所述通入二氧化碳后体系的压力为4~10mpa,例如4.5mpa、5mpa、5.5mpa、6mpa、6.5mpa、7mpa、7.5mpa、8mpa、8.5mpa、9mpa或9.5mpa等。
44.第四方面,本发明提供一种如第二方面所述的二氧化碳基共聚物在生物降解产品中的应用。
45.相对于现有技术,本发明具有以下有益效果:
46.(1)本发明提供复合催化剂包括非均相催化剂和环状酸酐的组合;所述复合催化剂可用于二氧化碳基共聚物的制备,且所述复合催化剂用于制备二氧化碳基共聚物时,由于环状酸酐的引入,可使制备得到的聚合物链增长的端基保持为无回咬的羧酸金属端基,取代了环氧化物和二氧化碳共聚形成的易回咬的羧基金属端基,从而大幅降低产物中副产物的生成量,提高了环氧化物的转化率以及二氧化碳基共聚物的选择性,具有重要研究意义。
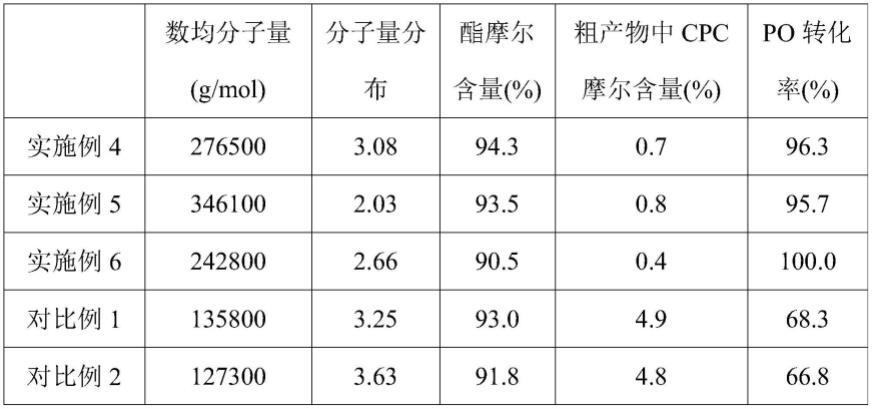
47.(2)具体而言,采用本发明提供的复合催化剂制备得到的二氧化碳基共聚物的聚合物分子量为242800~346100g/mol,分子量分布为2.03~3.08,酯摩尔含量为90.5~94.3%,粗产物中副产物cpc摩尔含量为0.4~0.81%,po单体转化率为96.3~100%。
附图说明
48.图1为实施例6得到的产物的1h-nmr谱图,
49.其中,1-氯仿中氢的特征峰,2-二氯甲烷中氢的特征峰,3-聚碳酸亚丙酯结构中的次甲基氢的特征峰和碳酸丙烯酯结构中次甲基氢的特征峰,4-碳酸丙烯酯结构中亚甲基氢
的特征峰,5-聚碳酸亚丙酯结构中亚甲基氢的特征峰和碳酸丙烯酯结构中亚甲基氢的特征峰,6-聚环氧丙烷结构中亚甲基氢和次甲基氢的特征峰,7-未反应的1,2-环己二甲酸临近羰基的次甲基氢的特征峰,8-参与反应的1,2-环己二甲酸酐临近羰基的次甲基氢的特征峰。
具体实施方式
50.下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
51.实施例1
52.一种复合催化剂,由甲基四氢邻苯二甲酸酐和稀土三元催化剂组成;
53.其中,稀土三元催化剂由0.75mmol的y(ccl3coo)3、7.5mmol甘油、15mmol的znet2和20ml的1,3-二氧五环组成,其具体制备方法参考cn1250603c;
54.本实施例提供的复合催化剂的制备方法包括:将30ml稀土三元催化剂和9g甲基四氢邻苯二甲酸酐在水浴中震荡混合1h,得到所述复合催化剂。
55.实施例2
56.一种复合催化剂,由戊二酸锌和邻苯二甲酸酐组成;
57.其中,戊二酸锌按照cn105418907a中实施例1制备得到;
58.本实施例提供的复合催化剂的制备方法包括:将2.67g戊二酸锌和4g邻苯二甲酸酐在研钵中研磨20min,通过400目筛网过滤,得到所述复合催化剂。
59.实施例3
60.一种复合催化剂,由zn-co双金属氰化物和1,2-环己二甲酸酐组成;
61.其中,zn-co双金属氰化物按照cn102432857a实施例1中提供的方法制备得到;
62.本实施例提供的复合催化剂的制备方法包括:称取0.133g的zn-co双金属氰化物加入13.8g的1,2-环己二甲酸酐,用研钵研磨20min,研磨后通过400目筛网过滤,得到所述复合催化剂。
63.实施例4
64.一种二氧化碳基共聚物,其制备方法包括如下步骤:
65.(1)将实施例1制备得到的复合催化剂投入经过无水无氧预处理的1l高压反应釜中,加入75ml环氧丙烷和55ml的1,3-二氧五环,充入co2使得温度90℃时釜内压力为8.0mpa,待聚合反应5h后,得到预聚物溶液;
66.(2)将125ml环氧丙烷与225ml的1,3-二氧五环的混合溶液于16h内匀速泵入步骤(1)得到的预聚物溶液中,待混合溶液泵入完毕,保持8.0mpa、90℃继续反应3h,得到初始产物;
67.(3)将循环水迅速切换成冷水,并在釜内盘管中通入冷却液,将体系温度迅速冷却至低于30℃,将反应釜内压力缓慢泄压至常压,停止搅拌,打开反应釜取出釜内混合产物,取适量混合产物样品进行1h-nmr测试,剩余样品用乙醇沉淀、洗涤,得到聚碳酸亚丙酯初产物,滤干后转移至真空烘箱内40℃、-0.1mpa处理10h,脱除残余的乙醇,得到281.4g纯净的聚碳酸亚丙酯。
68.实施例5
69.一种二氧化碳基共聚物,其制备方法包括如下步骤:
70.(1)将6.67g复合催化剂(实施例2)投入经过无水无氧预处理的1l高压反应釜中,加入75ml环氧丙烷和75ml四氢呋喃,充入co2使得温度90℃时釜内压力为8.0mpa,待聚合反应5h后,得到预聚物溶液;
71.(2)将125ml环氧丙烷与225ml的四氢呋喃的混合溶液于16h内匀速泵入步骤(1)得到的预聚物溶液中,待混合溶液泵入完毕,保持8.0mpa、90℃继续反应3h,得到初始产物;
72.(3)将循环水迅速切换成冷水,并在釜内盘管中通入冷却液,将体系温度迅速冷却至低于30℃,将反应釜内压力缓慢泄压至常压,停止搅拌,打开反应釜取出釜内混合产物,取适量混合产物样品进行1h-nmr测试,测试得到邻苯二甲酸酐转化率100%;剩余样品用乙醇沉淀、洗涤,得到聚碳酸亚丙酯初产物,滤干后转移至真空烘箱内40℃、-0.1mpa处理10h,脱除残余的乙醇,得到273.4g纯净的聚碳酸亚丙酯。
73.实施例6
74.一种二氧化碳基共聚物,其制备方法包括如下步骤:
75.(1)将13.933g复合催化剂(实施例3)投入经过无水无氧预处理的1l高压反应釜中,加入75ml环氧丙烷和75ml二氯甲烷,充入co2使得温度55℃时釜内压力为6.5mpa,待聚合反应3h后,得到预聚物溶液;
76.(2)将125ml环氧丙烷与225ml的二氯甲烷的混合溶液于12h内匀速泵入步骤(1)得到的预聚物溶液中,待混合溶液泵入完毕,保持6.5mpa、55℃继续反应3h,得到初始产物;
77.(3)将循环水迅速切换成冷水,并在釜内盘管中通入冷却液,将体系温度迅速冷却至低于30℃,将反应釜内压力缓慢泄压至常压,停止搅拌,打开反应釜取出釜内混合产物,取适量混合产物样品进行1h-nmr测试,剩余样品用乙醇沉淀、洗涤,得到聚碳酸亚丙酯初产物,滤干后转移至真空烘箱内40℃、-0.1mpa处理10h,脱除残余的乙醇,得到292.6g聚碳酸亚丙酯。
78.本实施例测试得到的1h-nmr谱图如图1所示,图1中,1代表氯仿中氢的特征峰,2代表二氯甲烷中氢的特征峰,3代表聚碳酸亚丙酯结构中的次甲基氢的特征峰和碳酸丙烯酯结构中次甲基氢的特征峰,4代表碳酸丙烯酯结构中亚甲基氢的特征峰,5代表聚碳酸亚丙酯结构中亚甲基氢的特征峰和碳酸丙烯酯结构中亚甲基氢的特征峰,6代表聚环氧丙烷结构中亚甲基氢和次甲基氢的特征峰,7代表未反应的1,2-环己二甲酸临近羰基的次甲基氢的特征峰,8代表参与反应的1,2-环己二甲酸酐临近羰基的次甲基氢的特征峰;从图1可以看出,没有出现原料po的特征峰,说明po的转化率很高,且聚碳酸亚丙酯结构中次甲基氢特征峰3和聚碳酸亚丙酯结构中亚甲基氢特征5峰十分明显,说明产物中主要为聚碳酸亚丙酯为主,而聚环氧丙烷结构中亚甲基氢和次甲基氢的特征峰6不是很明显,说明产物中的聚碳酸亚丙酯中的聚醚链段较少,图1中碳酸丙烯酯结构中亚甲基氢的特征峰4十分不明显,几乎没有,说明产物中的副产物碳酸丙烯酯的生成量很少;且进一步而言,参与反应的1,2-环己二甲酸酐临近羰基的次甲基氢的特征峰8相较于未反应的1,2-环己二甲酸临近羰基的次甲基氢的特征峰7更明显,说明酸酐大部分都参与了反应证明了酸酐参与反应,阻碍了回咬反应的发生。
79.对比例1
80.一种二氧化碳基共聚物,其制备方法包括如下步骤:
81.(1)将30ml稀土三元催化剂(由0.75mmol的y(ccl3coo)3、7.5mmol甘油、15mmol的znet2和20ml的1,3-二氧五环组成,其具体制备方法参考cn1250603c)投入经过无水无氧预处理的1l高压反应釜中,加入75ml环氧丙烷和55ml的1,3-二氧五环,充入co2使得温度90℃时釜内压力为8.0mpa,聚合反应5h后,得到初始产物的预聚物溶液;
82.(2)将125ml环氧丙烷与225ml的1,3-二氧五环的混合溶液于16h内匀速泵入步骤(1)得到的预聚物溶液中,待混合溶液泵入完毕,保持8.0mpa、90℃继续反应3h,得到初始产物;
83.(3)将循环水迅速切换成冷水,并在釜内盘管中通入冷却液,将体系温度迅速冷却至低于30℃,将反应釜内压力缓慢泄压至常压,停止搅拌,打开反应釜取出釜内混合产物,取适量混合产物样品进行1h-nmr测试,剩余样品用乙醇沉淀、洗涤,得到聚碳酸亚丙酯初产物,滤干后转移至真空烘箱内40℃、-0.1mpa处理10h,脱除残余的乙醇,得到193.8g聚碳酸亚丙酯。
84.对比例2
85.一种二氧化碳基共聚物,其制备方法包括如下步骤:
86.(1)将2g的znga(按照cn105418907a中实施例1提供的方法制备得到)投入经过无水无氧预处理的1l高压反应釜中,加入75ml环氧丙烷和75ml四氢呋喃,充入co2使得温度90℃时釜内压力为8.0mpa,聚合反应5h后,得到预聚物溶液;
87.(2)将125ml环氧丙烷与225ml的四氢呋喃的混合溶液于16h内匀速泵入步骤(1)得到的预聚物溶液中,待混合溶液泵入完毕,保持8.0mpa、90℃继续反应3h,得到初始产物;
88.(3)将循环水迅速切换成冷水,并在釜内盘管中通入冷却液,将体系温度迅速冷却至低于30℃,将反应釜内压力缓慢泄压至常压,停止搅拌,打开反应釜取出釜内混合产物,取适量混合产物样品进行1h-nmr测试,剩余样品用乙醇沉淀、洗涤,得到聚碳酸亚丙酯初产物,滤干后转移至真空烘箱内40℃、-0.1mpa处理10h,脱除残余的乙醇,得到188.4g聚碳酸亚丙酯。
89.对比例3
90.一种二氧化碳基共聚物,其制备方法包括如下步骤:
91.(1)将0.133g的zn-co双金属氰化物(按照cn102432857a实施例1提供的制备方法制备得到)投入经过无水无氧预处理的1l高压反应釜中,加入75ml环氧丙烷和75ml二氯甲烷,充入co2使得温度55℃时釜内压力为6.5mpa,聚合反应3h后,得到预聚物溶液;
92.(2)将125ml环氧丙烷与225ml的二氯甲烷的混合溶液于12h内匀速泵入步骤(1)得到的预聚物溶液中,待混合溶液泵入完毕,保持6.5mpa、55℃继续反应3h,得到初始产物;
93.(3)将循环水迅速切换成冷水,并在釜内盘管中通入冷却液,将体系温度迅速冷却至低于30℃,将反应釜内压力缓慢泄压至常压,停止搅拌,打开反应釜取出釜内混合产物,取适量混合产物样品进行1h-nmr测试,剩余样品用乙醇沉淀、洗涤,得到聚碳酸亚丙酯初产物,滤干后转移至真空烘箱内40℃、-0.1mpa处理10h,脱除残余的乙醇,得到263.8g聚碳酸亚丙酯。
94.性能测试:
95.(1)数均分子量和分子量分布:以窄分布聚苯乙烯为标样,二氯甲烷为流动相,采用gpc对产物进行测试;
96.(2)酯摩尔含量和碳酸丙烯酯(cpc)的摩尔含量:采用1h-nmr测试聚碳酸亚丙酯链段中酯摩尔含量以及粗产物中碳酸丙烯酯摩尔含量;酯摩尔含量计算公式为:酯摩尔含量=((a
5.0-a
4.6
)/(a
5.0
+a
3.5
/3))
×
100%,cpc含量计算公式为:cpc含量=(a
4.6
/(a
5.0
+a
4.6
+a
3.5
/3))
×
100%,其中,a表示1h-nmr谱图中对应化学位移的积分面积;
97.(3)po转化率:根据po和酸酐的投加质量、聚合物中酯含量、粗产物cpc含量和所得聚合物干重计算得到,计算公式为:po转化率=((实际所得聚碳酸亚丙酯质量-酸酐质量)/(po质量
×
(102
×
(1-cpc摩尔含量)
×
酯摩尔含量+58
×
(1-cpc摩尔含量)
×
(1-酯摩尔含量))))
×
100%。
98.按照上述测试方法对实施例4~6和对比例1~3得到的二氧化碳基共聚物进行测试,测试结果如表1所示:
99.表1
[0100][0101][0102]
根据表1可以看出:
[0103]
实施例4~6使用复配了酸酐的复合催化剂,使得到的二氧化碳基共聚物的分子量为242800~346100g/mol,分子量分布为2.03~3.08,酯摩尔含量为90.5~94.3%,粗产物中副产物cpc摩尔含量为0.4~0.81%,po单体转化率为96.3~100%。
[0104]
而在对比例1~3中,未使用复配酸酐的复合催化剂,进而得到的二氧化碳基共聚物的分子量均低于200000g/mol,酯摩尔含量和po转化率依据催化剂不同有所不同,但均低于实施例4~6的结果,且粗产物中副产物cpc含量均大于2%,部分催化剂高达5%左右。
[0105]
综上可以证明,本发明通过使用复配了酸酐的复合催化剂催化环氧化物和二氧化碳共聚,可以明显提高二氧化碳基共聚物的分子量、原料的转化率和碳酸酯的含量,同时还明显降低了粗产物中副产物的含量,有效提高了生产效率,降低了生产成本,提高了产品品质。
[0106]
申请人声明,本发明通过上述实施例来说明一种复合催化剂、二氧化碳基共聚物及其制备方法和应用,但本发明并不局限于上述工艺步骤,即不意味着本发明必须依赖上述工艺步骤才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围
和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1