一种负载型磁性纳米颗粒的Pickering界面有氧氧化反应的制作方法
一种负载型磁性纳米颗粒的pickering界面有氧氧化反应
技术领域
1.本发明涉及苯甲醛制备技术领域,具体涉及一种负载型磁性纳米颗粒的pickering界面有氧氧化反应。
背景技术:
2.苯甲醛是一种最简单的芳香醛,广泛的应用于香料、医药、染料和农药等行业。在工业生产中,苯甲醛通常是在碱性环境下通过水解氯化苄或者氧化甲苯来生产获得的。然而,在水解氯化苄的生产过程中,产物中往往会残留微量的氯,难以被去除;而对于甲苯氧化过程来说,产物虽无残留,但产物苯甲醛的选择性较低。
3.为了解决现有苯甲醛工业生产存在的上述问题,研究人员提出了无碱条件下通过有氧氧化苯甲醇制备苯甲醛的方法,即利用分子氧直接氧化苯甲醇为苯甲醛,在具体实施时分为气相氧化和液相氧化两种氧化方式。
4.气相氧化是指在固体催化剂及氧气作用下,汽化的苯甲醇进行气-固相催化氧化制备苯甲醛的连续反应。该方法易产生二氧化碳,不利于“双碳”目标的实现。
5.液相氧化是指在固体催化剂及氧气作用下,液态苯甲醇进行气-液-固相催化氧化制备苯甲醛的连续反应。基于环境、政策和经济的多重考虑,相比气相氧化,液相氧化具有反应温度低,不产生二氧化碳,苯甲醛选择性较高的优点。
6.然而,由于液相氧化体系为互不相容的气-液-固三相反应,反应过程中受到严重的传质影响。此外,液相氧化方式还存在溶剂毒性大、后处理难度大、催化剂重复使用活性下降等问题。
7.例如,alamgholiloo等[alamgholiloo h,rostamnia s,zhang k,et al.acs omega,2020,5(10):5182-5191.]合成了复合催化剂fe3o4/cu-bdc/go,将该催化剂用于苯甲醛的有氧氧化反应中,分别讨论了不同的溶剂、不同温度、不同酸碱性等条件对反应的影响,结果发现以乙腈为溶剂时,相比其他条件下反应具有更好的转化率和选择性,作者将原因归结为cu-mof与go、fe3o4以及2,2,6,6-四甲基哌啶氧化物的协同效应。但由于乙腈的高毒性,在试验过程和后续处理方面存在一定的问题,因此在经济性方面和环境影响方面均存在不利因素。
[0008]
hu等[hu z,zhou g,xu l,et al.applied surface science,2019,471:852-861.]合成了一系列pd/ceo
2-ng(氮掺杂的石墨烯),将该催化剂用于苯甲醇的有氧氧化反应中,结果表明:虽然该反应避免了使用有毒的乙腈溶剂,但是相对来说,该反应体系的反应效率相对较低,反应6h以后,反应的转化率的斜率大幅度减小,即说明了该反应的转化率趋于平稳,另外,对于回收重复使用性能方面,虽然反应后对苯甲醛的选择性基本维持不变,但是反应底物苯甲醇的转化率相对来说有大幅度的降低。
[0009]
为了解决苯甲醇液相有氧氧化存在的问题,现有技术引入了pickering界面反应的概念。由于pickering界面反应能够极大地降低互不相容相的表面张力,提高相与相之间的传质速率,从而提高反应的效率,降低反应的能耗,在“双碳”和节能降耗的背景下起到了
很强的推动作用。
[0010]
cn110483263a公开一种利用负载型钯催化剂催化氧化苯甲醇制备苯甲醛的方法,其【0005】记载了将钯盐与载体制成分散液,分散液在水-油两相中形成pickering乳液,在pickering乳液中钯盐氧化苯甲醇,同时自身还原并原位固载在界面处的载体上,生成负载型钯盐催化剂,并利用该催化剂进一步催化苯甲醇氧化生成苯甲醛。其【0011】段记载了该反应的反应条件温度、避免环境问题;所得催化剂可通过离心或过滤等简单操作回收,可多次重复使用;苯甲醇的转化率高,所得苯甲醛的选择性高,如实施例1所示。
[0011]
然而,由于该方法中的催化剂尺寸较小(纳米级)的原因,采用离心或过滤等常规分离方法难以将其与液相彻底分离,易造成催化剂流失的问题,进而导致重复使用后效率降低,须补充催化剂导致成本提高的问题,从而限制了pickering乳液反应的推广和应用。
技术实现要素:
[0012]
为了解决上述技术问题,本发明提供一种新的液相有氧氧化方法,该方法不仅具有较高的反应效率,而且催化剂易回收,重复使用效率高。
[0013]
本发明提供的液相有氧氧化方法是以表面具有超疏水性的、负载贵金属的sio2@fe3o4纳米颗粒为催化剂进行反应。
[0014]
本发明基于反应物的特性,不仅在常压条件下将pickering界面反应引入到固-液-气三相反应中,而且利用耦合pickering界面反应和超顺磁性特点的催化剂,显著提高了液相有氧氧化反应效率及催化剂重复使用的效率。
[0015]
具体来讲,含有该催化剂的反应体系在通入氧气及剧烈搅拌条件下会形成各种泡沫,这可延长氧气在反应体系中的停留时间,实现深度氧化,因此在相同反应时间内,利用该催化剂呈现出更好的pickering界面催化反应效果(如实施例),间接地降低能耗,为固-液-气三相反应开辟一种新颖的反应路径。
[0016]
同时,本发明利用四氧化三铁的超顺磁性,在外磁场作用下更为容易地将催化剂彻底分离出来,同时又可在撤去磁场的条件下,经过简单的搅拌使其重新均匀的分散于溶液中,并经简单洗涤干燥后即可实现催化剂再生;而且由于整个过程中催化剂几乎不产生流失,再生后的催化剂的活性基本保持不变,避免了现有固-液-气三相反应分离回收时催化剂易流失和难回收的问题;不但可以减少反应后处理繁琐的操作,而且也符合“双碳”和节能降耗的大背景。
[0017]
进一步地,所述催化剂通过下述方法制得:
[0018]
(1)在含有fe3o4纳米颗粒、硅酸乙酯的体系中,于碱性条件下水解硅酸乙酯,得到二氧化硅壳层包覆的四氧化三铁纳米颗粒,即磁性sio2@fe3o4;
[0019]
(2)利用硅烷偶联剂改性所得磁性sio2@fe3o4,得到表面修饰的具有超疏水性的sio2@fe3o4;
[0020]
(3)对所得表面修饰的具有超疏水性的sio2@fe3o4负载贵金属。
[0021]
相比现有利用硅酸钠及模板剂合成sio2壳层,本发明通过碱性水解硅酸乙酯制备sio2壳层的方式,省略了焙烧去除模板剂的步骤,简化制备工艺,并且在合成过程中省去了过滤、干燥等步骤,直接在表面修饰上了超疏水性的基团,大大节省了合成所用的时间和能耗;在此基础上,本发明利用硅烷偶联剂对其改性,使其表面呈现超疏水性,从而可在后续
液相有氧氧化反应中,利用氧气及剧烈搅拌形成各种泡沫,显著延长氧气停留时间,进而提高反应效率。
[0022]
所述步骤(2)中,所述硅烷偶联剂选自1h,1h,2h,2h-全氟癸基三乙氧基硅烷、1h,1h,2h,2h-全氟辛基三乙氧基硅烷、辛烷基三乙氧基硅烷、3,3,3-三氟丙基三甲氧基硅烷。
[0023]
优选地,所述硅烷偶联剂为1h,1h,2h,2h-全氟癸基三乙氧基硅烷,相比其他硅烷偶联剂,其具有更长的碳链,更多的氟原子,因此具有更强的疏水性的优势,以便后续更容易的形成泡沫,从而提高反应的效率。
[0024]
所述硅烷偶联剂与硅酸乙酯的摩尔比为2:1~1:8;优选地,所述硅烷偶联剂与硅酸乙酯的摩尔比为1:4。研究表明,硅酸乙酯的含量过高会导致在普通磁场的分离作用下速度降低,而含量过少又存在包覆不均匀的问题,通过控制二者合适的摩尔比例,实现在在普通磁场下即可简单分离的目的。
[0025]
所述改性的反应温度为40-42℃,反应时间为3-3.5h。
[0026]
所述步骤(1)中,所述反应的体系ph控制在8.9-9.1。通过控制体系的ph,有助于水解更彻底,所形成的sio2壳层包覆更均匀。
[0027]
所述硅酸乙酯与fe3o4纳米颗粒的用量比例为(1.5-2)ml:1g。
[0028]
所述步骤(3)中,所述负载具体过程为:将表面修饰的具有超疏水性的sio2@fe3o4添加到含有贵金属盐的有机溶液中,分散后冷却,再利用还原剂还原贵金属,使其负载纳米颗粒表面。
[0029]
相比现有贵金属负载采用超声方式将贵金属组装在介孔sio2壳层的负载方式,本发明利用还原剂将贵金属还原并负载于纳米颗粒表面的方式具有反应时间短,反应条件温和、而且在溶液中即可将钯盐原位还原的优势,省去了后续氢气还原或者其他方式还原带来的不便。
[0030]
所述贵金属盐为醋酸钯、硝酸钯、氯化钯等钯盐;
[0031]
所述还原剂为硼氢化钾、硼氢化钠、水合肼等。
[0032]
进一步地,本发明还提供了利用上述方法制备苯甲醛。
[0033]
本发明所述的苯甲醛的制备方法,是采用上述催化剂,通过苯甲醇的液相有氧氧化反应制得的。
[0034]
所述液相有氧氧化反应中,所述催化剂的用量为底物苯甲醇质量的1.0wt%;氧气的通入速度为40-45ml/min,反应温度为120-125℃,搅拌速度为1500-1600转/分。本发明利用各反应物及催化剂的特性,在上述条件下形成大量泡沫,延长了氧气的保留时间,增加了彼此之间的相互接触。
[0035]
本发明取得的有益效果如下:
[0036]
(1)本发明充分利用反应物的特性,在常压的条件下,将耦合了超顺磁性、表面超疏水性和pickering界面反应的优点的催化剂引入苯甲醇液相氧化制备苯甲醛的反应中,显著提高了液-固-气三相反应效率;该方法具有一定的普适性,可以较为容易的推广到其他相关的液-固-气三相反应中。
[0037]
(2)从催化剂回收利用的角度来看,本发明所述催化剂呈现超顺磁性,在回收重复使用方面具有反应后分离容易,并且几乎不会产生流失;而在撤去磁场的条件下,经过简单的搅拌该催化剂又可以均匀的分散在溶液中,催化剂回收后后续经过简单的洗涤干燥,催
化剂即可以再生,无须其他复杂的手段;而再生之后,催化剂的活性基本保持不变,避免了固-液-气三相反应分离回收时,催化剂易流失和难回收的问题,从而也就避免了催化剂活性降低的问题。
[0038]
(3)从反应过程强化的角度来看,本发明所述催化剂具有表面超疏水性和pickering界面反应的优点,在通入氧气及剧烈搅拌条件下形成泡沫,延长了氧气在反应体系中的停留时间,增加反应物相互接触,因此在相同反应时间内,显著提高苯甲醇的转化率。
附图说明
[0039]
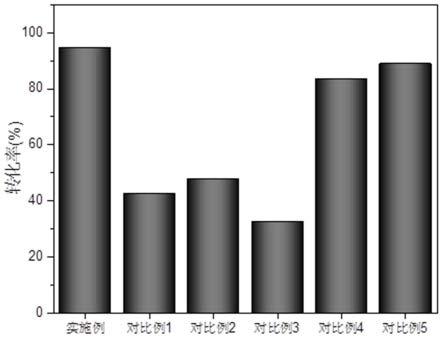
图1为实施例及对比例中苯甲醇的转化率。
具体实施方式
[0040]
以下实施例用于说明本发明,但不用来限制本发明的范围。
[0041]
实施例
[0042]
本实施例提供一种催化剂及苯甲醛的制备方法,具体步骤如下:
[0043]
(1)催化剂的制备:
[0044]
s1、7.5g醋酸钠和2.7g三氯化铁溶解于150ml乙二醇中,然后转移至不锈钢反应釜,于200℃搅拌12h,冷却磁性分离,用乙醇和去离子水清洗后真空干燥,得到磁性四氧化三铁纳米颗粒。
[0045]
s2、随后按照0.1g四氧化三铁分散在100ml乙醇(乙醇和去离子水的体积比为4:1)溶液中,控制ph值为9左右,滴加0.15ml硅酸乙酯teos,继续搅拌6h,得到磁性二氧化硅包覆四氧化三铁sio2@fe3o4纳米颗粒;
[0046]
在氮气保护的条件下,继续添加硅烷偶联剂1h,1h,2h,2h-全氟癸基三乙氧基硅烷(teos与1h,1h,2h,2h-全氟癸基三乙氧基硅烷的摩尔比为4:1),置于40℃条件下继续搅拌3h,然后经分离、洗涤和真空干燥后得到表面修饰的磁性二氧化硅包覆四氧化三铁。
[0047]
s3、然后按照0.3g表面修饰的磁性二氧化硅包覆四氧化三铁添加到20ml醋酸钯的乙醇溶液(浓度为1.0g/l)中,充分分散后,置于冰水混合物中,再用硼氢化钾溶液还原,继续搅拌3h后,离心分离、清洗、真空干燥得负载pd的表面修饰的磁性二氧化硅包覆四氧化三铁,标记为pd/sio2@fe3o4(f-17)。
[0048]
(2)苯甲醛的制备:
[0049]
将上述所合成的pd/sio2@fe3o4(f-17)催化剂1.0wt%加入到1.8ml苯甲醇(反应底物)、1.8ml二甲苯(溶剂)的混合溶液中,再以40ml/min的氧气流速通入到上述混合液中,然后在120℃,1500转/分条件下搅拌1h。
[0050]
反应结束后,添加5ml丙酮和0.1g内标物联苯,磁性分离,取上清液进行色谱分析,计算苯甲醇的转化率为94.7%。
[0051]
(3)催化剂的回收:
[0052]
并且将该催化剂回收清洗干燥(80℃)后,重新用于上述反应,重复使用五次,催化剂的活性仍保持在91%左右。
[0053]
对比例1(以fe3o4为催化剂)
[0054]
7.5g醋酸钠和2.7g三氯化铁溶解于150ml乙二醇中,然后转移至不锈钢反应釜,于200℃搅拌12h,冷却磁性分离,用乙醇和去离子水清洗后真空干燥,得到四氧化三铁纳米颗粒。
[0055]
将上述所合成的fe3o4催化剂1.0wt%加入到1.8ml苯甲醇(反应底物)、1.8ml二甲苯(溶剂)的混合溶液中,再以40ml/min的氧气流速通入到上述混合液中,然后在120℃,1500转/分条件下搅拌1h。反应结束后,添加5ml丙酮和0.1g联苯作为内标物,磁性分离,取上清液进行色谱分析,计算苯甲醇的转化率为42.6%。
[0056]
对比例2(以未负载贵金属、表面修饰的sio2@fe3o4为催化剂)
[0057]
7.5g醋酸钠和2.7g三氯化铁溶解于150ml乙二醇中,然后转移至不锈钢反应釜,于200℃搅拌12h,冷却磁性分离,用乙醇和去离子水清洗后真空干燥,得到四氧化三铁纳米颗粒。
[0058]
随后按照0.1g四氧化三铁分散在100ml乙醇(乙醇和去离子水的体积比为4:1)溶液中,控制ph值为9左右,滴加0.15ml teos,继续搅拌6h后在氮气保护的条件下,添加1h,1h,2h,2h-全氟癸基三乙氧基硅烷(teos与1h,1h,2h,2h-全氟癸基三乙氧基硅烷的摩尔比为4:1),置于40℃条件下继续搅拌3h,然后经分离、洗涤和真空干燥后得到表面修饰的磁性二氧化硅包覆四氧化三铁。
[0059]
将上述所合成的表面修饰的sio2@fe3o4(f-17)催化剂1.0wt%加入到1.8ml苯甲醇(反应底物)、1.8ml二甲苯(溶剂)的混合溶液中,再以40ml/min的氧气流速通入到上述混合液中,然后在120℃,1500转/分条件下搅拌1h。反应结束后,添加5ml丙酮和0.1g内标物联苯,磁性分离,取上清液进行色谱分析,计算苯甲醇的转化率为47.9%。
[0060]
对比例3(以未负载贵金属、未表面修饰的sio2@fe3o4为催化剂)
[0061]
7.5g醋酸钠和2.7g三氯化铁溶解于150ml乙二醇中,然后转移至不锈钢反应釜,于200℃搅拌12h,冷却磁性分离,用乙醇和去离子水清洗后真空干燥,得到四氧化三铁纳米颗粒。
[0062]
随后按照0.1g四氧化三铁分散在100ml乙醇(乙醇和去离子水的体积比为4:1)溶液中,控制ph值为9左右,滴加0.15ml teos,继续搅拌6h后经分离、洗涤和真空干燥后得到磁性二氧化硅包覆四氧化三铁纳米颗粒,即sio2@fe3o4催化剂。
[0063]
将上述所合成的sio2@fe3o4催化剂1.0wt%加入到1.8ml苯甲醇(反应底物)、1.8ml二甲苯(溶剂)的混合溶液中,再以40ml/min的氧气流速通入到上述混合液中,然后在120℃,1500转/分条件下搅拌1h。反应结束后,添加5ml丙酮和0.1g内标物联苯,磁性分离,取上清液进行色谱分析,计算苯甲醇的转化率为32.7%。
[0064]
对比例4(以未表面修饰的sio2@fe3o4为催化剂)
[0065]
7.5g醋酸钠和2.7g三氯化铁溶解于150ml乙二醇中,然后转移至不锈钢反应釜,于200℃搅拌12h,冷却磁性分离,用乙醇和去离子水清洗后真空干燥,得到四氧化三铁纳米颗粒。
[0066]
随后按照0.1g四氧化三铁分散在100ml乙醇(乙醇和去离子水的体积比为4:1)溶液中,控制ph值为9左右,滴加0.15ml teos,继续搅拌6h后经分离、洗涤和真空干燥后得到磁性二氧化硅包覆四氧化三铁。
[0067]
然后按照0.3g表面修饰的磁性二氧化硅包覆四氧化三铁添加到20ml醋酸钯的乙
醇溶液(浓度为1.0g/l)中,充分分散后,置于冰水混合物中,再用硼氢化钾溶液还原,继续搅拌3h后,离心分离、清洗、真空干燥得负载pd的表面修饰的磁性二氧化硅包覆四氧化三铁,标记为pd/sio2@fe3o4。
[0068]
将上述所合成的pd/sio2@fe3o4催化剂1.0wt%加入到1.8ml苯甲醇(反应底物)、1.8ml二甲苯(溶剂)的混合溶液中,再以40ml/min的氧气流速通入到上述混合液中,然后在120℃,1500转/分条件下搅拌1h。反应结束后,添加5ml丙酮和0.1g内标物联苯,磁性分离,取上清液进行色谱分析,计算苯甲醇的转化率为83.6%。
[0069]
对比例5
[0070]
与实施例的区别在于,所述硅烷偶联剂为3,3,3-三氟丙基三甲氧基硅烷。
[0071]
结果显示,苯甲醇的转化率为88.9%,催化剂重复使用五次后活性为85%。
[0072]
上述实施例及对比例的转化率如图1所示。
[0073]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1