1.本发明涉及一种二元糖结晶及其制备方法。
背景技术:2.二元糖(也称为双糖或二糖)是由两分子单糖缩合生成(两个单糖通过糖苷键连接时形成的糖),或由多糖在酶的帮助下通过水解生成,1分子二元糖彻底水解后,只水解成2分子的单糖。与单糖一样,二糖是可溶于水的糖。常见的二糖有蔗糖、海藻糖、乳糖和麦芽糖。其化学组成具有12个碳原子,其通式为c
12h22o11
。
3.天然存在的游离态和具有机能的二元糖以麦芽糖、乳糖、海藻糖、蔗糖为代表。这些糖是作为各种生物体的能量来源或作为生物体组成的重要物质,其承担着贮藏或运输的重要作用。现有技术中有很多二元糖结晶的制备方法,但是现有的工艺中,二元糖结晶的生产周期长、产率低、纯度不高。
4.专利号cn107447058公开了一种结晶麦芽糖的制备方法,在该方法中并没有指出所用麦芽糖浆的麦芽糖含量,而实际情况是:如果糖浆中麦芽糖含量太低的话,结晶过程中容易出现糖膏粘稠,无法结晶现象。
5.专利号cn110938715a公开了一种麦芽糖结晶工艺,该工艺过程需要真空条件且结晶过程中降温速率缓慢,其结晶时间约38小时以上,生产周期较长,不适合工业化生产。
技术实现要素:6.本发明为了克服现有技术中二元糖结晶的生产周期长、产率低、纯度不高等缺陷,提供了一种二元糖结晶及其制备方法,其制备方法提高了二元糖晶体的收率和纯度,结晶时间大幅缩短,提高了生产效率;其制备得到的二元糖结晶具有颜色浅,纯度高,杂质少,细菌内毒素低的优点。
7.为了实现上述目的,本发明通过以下技术方案实现:
8.本发明提供了一种二元糖结晶的制备方法,其包括以下步骤:
9.s1、将二元糖溶液过滤、微滤、超滤,减压浓缩,得浓糖液;其中,所述二元糖溶液包括二元糖原料和水;所述超滤的分子截留量为1000kd~10000kd;
10.s2、将所述浓糖液和乙醇混合,降温,加入晶种后保温;所述乙醇和所述浓糖液的体积比为(0.4~0.6):1;所述晶种为二元糖晶体;
11.s3、降温,在降温过程中和乙醇混合,所述乙醇和所述浓糖液的体积比为(0.4~0.6):1。
12.步骤s1中,所述二元糖原料可为本领域常规,优选为食品级二元糖粉末或结晶。所述的二元糖较佳地为蔗糖、海藻糖、乳糖或麦芽糖。所述二元糖原料的纯度优选为大于92%,所述二元糖原料的纯度是指所述二元糖占所述二元糖原料的质量百分比。所述水优选为纯化水。
13.步骤s1中,所述二元糖占所述二元糖溶液的质量百分比优选为30%~50%,例如
40%。
14.步骤s1中,所述二元糖溶液可通过本领域常规制备方法得到,可将所述二元糖原料溶解在水中;所述二元糖原料的溶解温度优选为50-80℃,例如60℃。
15.其中,所述二元糖溶液的制备,在所述溶解后,较佳地,还包括脱色处理。
16.其中,所述脱色处理可采用本领域常规脱色方法,优选为采用药用活性炭粉进行所述脱色处理;所述药用活性炭粉的质量为所述二元糖溶液质量的0.05%~1%。所述脱色处理的温度优选为65~70℃,例如60℃。所述脱色处理的时间优选为40~60min,例如30min。
17.步骤s1中,所述过滤是指过滤精度大于50μm的过滤方法;所述微滤是指过滤精度在0.1~50μm的过滤方法;所述超滤是指过滤精度在0.001~0.1μm的过滤方法。过滤、微滤可除去较大机械杂质。
18.步骤s1中,所述超滤可采用超滤膜进行;所述超滤的分子截留量为,更优选为1000kd~5000kd。所述超滤膜的进口端压力优选为0.1~5bar。所述超滤膜的回流端压力优选为0.1~3bar。所述超滤膜的材料可为本领域常规,优选为改性聚砜。所述超滤的温度优选为20~35℃。
19.本发明中,所述改性聚砜是聚砜、聚甲基丙烯酸甲酯、abs的共混物;所述abs是丙烯腈、丁二烯、苯乙烯三种单体的三元共聚物。
20.步骤s1中,所述减压浓缩的温度优选为65~70℃。
21.步骤s1中,所述减压浓缩后,所述二元糖占所述浓糖液的质量百分比优选为65%~80%,例如70%、75%。
22.步骤s1中,所述混合的温度优选保持在50~65℃,例如55℃。
23.步骤s2中,所述混合的方式优选为在所述浓糖液中加入乙醇;所述乙醇的加入的速率为80~180ml/min。
24.步骤s2中,所述乙醇与所述浓糖液的体积比优选为0.55:1、0.50:1或0.45:1。
25.步骤s2中,所述混合的温度可为65~70℃。
26.步骤s2中,所述降温可采用本领域常规的降温方法,所述降温可降温至45~60℃,例如50℃、55℃。
27.步骤s2中,所述保温的时间优选为2~5小时,例如3小时。
28.步骤s2中,所述二元糖晶种可为本领域常规,优选为食品级二元糖晶体或者含量为92%~100%的二元糖晶体。
29.步骤s2中,所述二元糖晶种与所述二元糖原料的质量比优选为0.1%~0.8%,例如0.2%。
30.步骤s3中,所述降温可采用本领域常规降温方法;所述降温可降温至10~30℃,例如20℃。
31.步骤s3中,所述降温的速率优选为1~10℃/h,例如5℃/h。
32.步骤s3中,所述乙醇与所述浓糖液的体积比优选为0.45:1、0.55:1或0.50:1。
33.步骤s2和s3均可在本领域常规容器内进行,优选为真空结晶器。
34.优选地,步骤s3后还可包括分离、干燥。
35.其中,所述分离可采用本领域常规分离方法,优选地,可采用离心或过滤。所述分
离的温度优选在20℃以下时进行。
36.其中,所述干燥可采用本领域常规干燥方法,优选为在烘箱中干燥。所述干燥的温度可为80℃。
37.本发明提供了一种二元糖结晶,其由上述的二元糖结晶的制备方法制备得到,较佳地,所述二元糖结晶的细菌内毒素低于2eu/g,例如低于0.52eu/g。
38.上述各优选条件,可任意组合,即得本发明各较佳实例。
39.本发明所用试剂和原料均市售可得。
40.本发明的积极进步效果在于:
41.1.本发明使用超滤工艺降低了二元糖浆中的细菌内毒素和多糖杂质,通过降温结晶和反溶剂结晶相结合的工艺,以及在降温结晶过程中补加不良溶剂的方法缩短了二元糖结晶的时间,使糖液的饱和度趋于稳定。
42.2.采用本发明的方法得到的二元糖结晶具有颜色浅,纯度高,杂质少,细菌内毒素低的优点,其产品质量达到了2020年版中国药典、usp43版美国药典,jp17版日本药典的标准,具体为制得的二元糖产品含量达到了99%以上。所得二元糖产品细菌内毒素低于0.5eu/g,可满足高风险(如注射,生物制品等)药用辅料使用要求。
43.3.本发明的二元糖结晶的制备方法反应温度低,反应条件温和,结晶时间大幅缩短,结晶工艺时长可控制在12小时左右,缩短生产周期,提高了生产效率;收率可达80%以上;设备简单易于工业化和大批量生产。
附图说明
44.图1为效果实施例3对照品3的hplc色谱图。
45.图2为效果实施例3供试品3的hplc色谱图。
具体实施方式
46.下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
47.实施例1
48.步骤1、取纯度大于92%的食品级麦芽糖结晶200g;
49.步骤2、将步骤1中麦芽糖结晶溶于200g纯化水,得到麦芽糖溶液;
50.步骤3、加入0.2g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色处理30min;
51.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为1000kd,进口端压力为4.2bar,回流端压力为2.5bar;超滤温度为20℃;
52.步骤5、将超滤后的麦芽糖溶液于65℃下减压浓缩至质量分数为65%,得到浓糖液;
53.步骤6、向步骤5的浓糖液中缓慢加入45g乙醇,并降温至50℃,此时加入0.2g麦芽糖晶体作为晶种,之后保温2小时;乙醇的加入的速率为80ml/min;
54.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加55g乙醇;
55.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产
品麦芽糖结晶,收率为83.8%,含量为99.9%,内毒素限度为0.5eu。
56.实施例2
57.步骤1、取纯度大于92%的食品级海藻糖结晶200g;
58.步骤2、将步骤1中海藻糖结晶溶于200g纯化水,得到海藻糖溶液;
59.步骤3、加入0.4g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色30min;
60.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为5000kd,进口端压力为1.2bar,回流端压力为0.5bar;超滤温度为25℃;
61.步骤5、将超滤后的海藻糖溶液于70℃减压浓缩至质量分数为70%,得到浓糖液;
62.步骤6、向步骤5的浓糖液中缓慢加入40g乙醇,并降温至55℃,此时加入0.4g海藻糖晶体作为晶种,之后保温2小时;乙醇的加入的速率为120ml/min;
63.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加60g乙醇;
64.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产品海藻糖结晶,收率为82.1%,含量为99.9%,内毒素限度为1eu。
65.实施例3
66.步骤1、取纯度大于92%的食品级蔗糖结晶200g;
67.步骤2、将步骤1中蔗糖结晶溶于200g纯化水,得到蔗糖溶液;
68.步骤3、加入1g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色处理30min;
69.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为10000kd,进口端压力为3.2bar,回流端压力为2.0bar;超滤温度为35℃;
70.步骤5、将超滤后的蔗糖溶液于65℃减压浓缩至质量分数为75%,得到浓糖液;
71.步骤6、向步骤5的浓糖液中缓慢加入50g乙醇,并降温至60℃,此时加入0.2g蔗糖晶体作为晶种,之后保温2小时;乙醇的加入的速率为180ml/min;
72.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加50g乙醇;
73.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产品蔗糖结晶,收率为81.3%,含量为99.9%,内毒素限度为2eu。
74.对比例1
75.步骤1、取纯度大于92%的食品麦芽糖结晶200g;
76.步骤2、将步骤1中麦芽糖结晶溶于200g纯化水,得到麦芽糖溶液;
77.步骤3、加入0.2g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色处理30min;
78.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为1000kd,进口端压力为4.2bar,回流端压力为2.5bar;超滤温度为20℃;
79.步骤5、将超滤后的麦芽糖溶液于65℃减压浓缩至质量分数为65%;
80.步骤6、向步骤5的浓糖液中加入0.2g麦芽糖晶体作为晶种,之后保温2小时;
81.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加100g乙醇;乙醇的加入的速率为80ml/min;
82.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产品麦芽糖结晶,收率为79.8%,含量为97.3%,内毒素限度为0.5eu。
83.对比例2
84.步骤1、取纯度大于92%的食品级麦芽糖结晶200g;
85.步骤2、将步骤1中麦芽糖结晶溶于200g纯化水,得到麦芽糖溶液;
86.步骤3、加入0.2g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色处理30min;
87.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为1000kd,进口端压力为4.2bar,回流端压力为2.5bar;超滤温度为20℃;
88.步骤5、将超滤后的麦芽糖溶液于65℃减压浓缩至质量分数为65%;
89.步骤6、向步骤5的浓糖液中缓慢加入25g乙醇,并降温至50℃,此时加入0.2g麦芽糖晶体作为晶种,之后保温2小时;乙醇的加入的速率为80ml/min;
90.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加75g乙醇;
91.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产品麦芽糖结晶,收率为79.5%,含量为97.2%,内毒素限度为0.5eu。
92.对比例3
93.步骤1、取纯度大于92%的食品级麦芽糖结晶200g;
94.步骤2、将步骤1中麦芽糖结晶溶于200g纯化水,得到麦芽糖溶液;
95.步骤3、加入0.2g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色处理30min;
96.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为1000kd,进口端压力为4.2bar,回流端压力为2.5bar;超滤温度为20℃;
97.步骤5、将超滤后的麦芽糖溶液于65℃减压浓缩至质量分数为65%;
98.步骤6、向步骤5的浓糖液中缓慢加入70g乙醇,并降温至50℃,此时加入0.2g麦芽糖晶体作为晶种,之后保温2小时;乙醇的加入的速率为80ml/min;
99.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加30g乙醇;
100.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产品麦芽糖结晶,收率为80.4%,含量为97.3%,内毒素限度为0.5eu。
101.对比例4
102.步骤1、取纯度大于92%的食品级麦芽糖结晶200g;
103.步骤2、将步骤1中麦芽糖结晶溶于200g纯化水,得到麦芽糖溶液;
104.步骤3、加入0.2g活性炭粉,搅拌升温至60℃后,继续搅拌,脱色处理30min;
105.步骤4、过滤,微滤、超滤,其中超滤膜分子截流量为20000kd,进口端压力为4.2bar,回流端压力为2.5bar;超滤温度为20℃;
106.步骤5、将超滤后的麦芽糖溶液于65℃下减压浓缩至质量分数为65%,得到浓糖液;
107.步骤6、向步骤5的浓糖液中缓慢加入45g乙醇,并降温至50℃,此时加入0.2g麦芽糖晶体作为晶种,之后保温2小时;乙醇的加入的速率为80ml/min;
108.步骤7、之后以5℃/h的速率缓慢降温至20℃,并在此过程中滴加55g乙醇;
109.步骤8、将步骤7的反应液离心,将得到的固体置于80℃烘箱中干燥4h得到最终产品麦芽糖结晶,收率为84.1%,含量为98.3%,内毒素限度为5eu。
110.将实施例1-3和对比例1-4制备得到的二元糖结晶采用hplc高效液相色谱仪进行含量检测,hplc检测的含量计算公式为:
111.含量=(ru/rs)
×
(cs/cu)
×
(c1/c2)
×
100%
112.式中:ru:供试品的峰面积;
113.rs:对照品的峰面积;
114.cu:供试品的浓度(mg/ml);
115.cs:对照品的浓度(mg/ml);
116.c1:对照品的纯度;
117.c2:供试品的纯度。
118.采用实施例1-3和对比例1-4制备得到的二元糖结晶配制成供试品1、2、3、4、5、6和7,供试品1-7的浓度均约为5.02%。
119.效果实施例1
120.对实施例1和对比例1-4的供试品1和供试品4-7分别进行含量检测,其采用的对照品为对照品1。
121.对照品1:纯度为94.4%的麦芽糖结晶(中国食品药品检定研究院),对照品浓度为5.07mg/ml
122.色谱条件为:
123.流动相:乙腈-水,乙腈占流动相的体积百分比为70%
124.检测时间:30min
125.色谱柱:氨基键合硅胶为填充剂
126.柱温:35℃
127.流速:0.8ml/min
128.检测器:蒸发光散射检测器
129.漂移管温度:80℃
130.载气流速:1.5l/min
131.对照品1的峰面积和保留时间如下表1所示。
132.表1.对照品1的峰面积和保留时间数据
[0133][0134]
供试品1(实施例1)的峰面积和保留时间如下表2所示。
[0135]
表2.供试品1的峰面积和保留时间数据
[0136]
[0137]
供试品1的水分:5.5%
[0138]
供试品1(即实施例1)的麦芽糖结晶的含量计如下:
[0139][0140]
供试品4-7的水分:5.5%
[0141]
供试品4(对比例1)的峰面积和保留时间如下表3所示。
[0142]
表3.供试品4的峰面积和保留时间数据
[0143][0144]
供试品5(对比例2)的峰面积和保留时间如下表4所示。
[0145]
表4.供试品5的峰面积和保留时间数据
[0146][0147]
供试品6(对比例3)的峰面积和保留时间如下表5所示。
[0148]
表5.供试品6的峰面积和保留时间数据
[0149][0150]
供试品7(对比例4)的峰面积和保留时间如下表6所示。
[0151]
表6.供试品7的峰面积和保留时间数据
[0152][0153]
效果实施例2
[0154]
对实施例2的供试品2进行含量检测,其采用的对照品为对照品2。
[0155]
对照品2:纯度为99.7%的海藻糖结晶(中国食品药品检定研究院),对照品浓度为5.06g/ml
[0156]
色谱条件:
[0157]
流动相:水
[0158]
检测时间:30min
[0159]
色谱柱:磺化交联的苯乙烯-二乙烯基苯共聚物填充剂
[0160]
柱温:80℃
[0161]
流速:0.4ml/min
[0162]
检测器:示差折光检测器
[0163]
检测器温度:40℃
[0164]
对照品2的峰面积和保留时间如下表7所示。
[0165]
表7.对照品2的峰面积和保留时间数据
[0166][0167]
供试品2的水分:9.5%
[0168]
供试品2(实施例2)的峰面积和保留时间如下表8所示。
[0169]
表8.供试品2的峰面积和保留时间数据
[0170][0171]
效果实施例3
[0172]
对实施例3的供试品3进行含量检测,其采用的对照品为对照品3。
[0173]
对照品3:纯度为100%的蔗糖结晶(中国食品药品检定研究院),对照品浓度为5.02mg/ml。
[0174]
色谱条件:
[0175]
流动相:水
[0176]
检测时间:30min
[0177]
色谱柱:糖分析柱
[0178]
柱温:60℃
[0179]
流速:0.5ml/min
[0180]
检测器:示差折光检测器
[0181]
检测器温度:40℃
[0182]
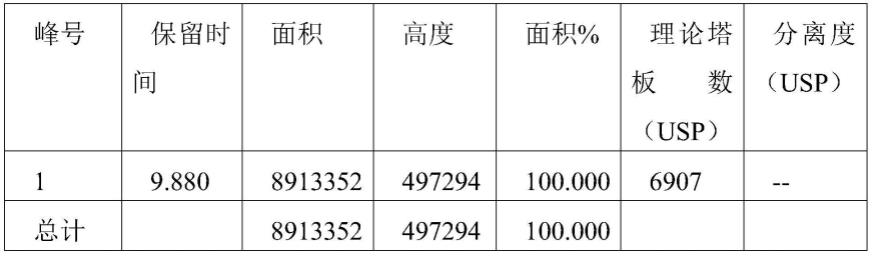
图1为对照品3的hplc色谱图,其中峰面积和保留时间如下表9所示。
[0183]
表9.对照品3的峰面积和保留时间数据
[0184][0185][0186]
供试品3的水分:0.3%
[0187]
图2为供试品3(实施例3)的hplc色谱图,其中峰面积和保留时间如下表10所示。
[0188]
表10.供试品3的峰面积和保留时间数据
[0189]