一种高效制备孟鲁司特钠侧链中间体的方法与流程
1.本发明涉及生物制药技术领域,尤其涉及一种孟鲁司特钠侧链中间体的制备工艺。
背景技术:
2.孟鲁司特钠,是一种平喘抗炎和抗过敏药,用于哮喘的预防和长期治疗,包括预防白天和夜间的哮喘症状,治疗对阿斯匹林敏感的哮喘患者以及预防运动诱发的支气管收缩。其化学名称是1-[[[(1r)-1-[3-[(1e)-2-(7-氯-2-喹啉)乙烯基]苯基]-3-[2-(1-羟基-1-甲基乙基)苯基]丙基]硫代]甲基]环丙烷乙酸钠,结构式如下:
[0003][0004]
通过分析其结构式可知,侧链部分的1-(巯基甲基)环丙基乙酸或其衍生物是合成孟鲁司特钠的关键中间体,1-羟甲基环丙基乙腈又是合成1-(巯基甲基)环丙基乙酸的一个关键中间体。
[0005]
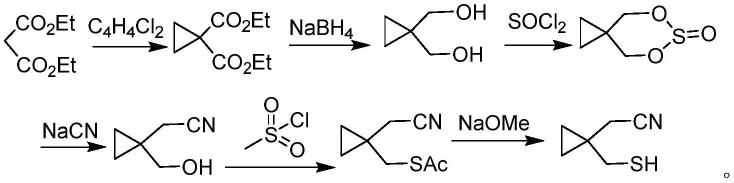
目前,关于合成1-巯甲基环丙基乙腈的报道不多,专利wo2008058118a2公开了一种1-巯甲基环丙基乙腈的制备工艺:以丙二酸二乙酯为起始原料,经过环合、还原制得1,1-环丙基二甲醇,再依次经过亚硫酰氯、氰化钠、甲烷磺酰氯、甲醇钠的作用,经过四步反应合成目标产物。具体路线如下所示:
[0006][0007]
该方法的工艺路线较长,整个过程繁琐。
[0008]
另一种是用1-羟甲基环丙基乙腈为原料,分别与溴化物、硫脲、碱、金属发生反应,合成1-巯甲基环丙基乙酸。该制备方法工艺路线长,且该制备方法需在100℃条件下反应12h,温度较高且时间长,难免发生副反应,生成副产物,以致1-巯甲基环丙基乙酸产品纯度低。如路线(1)所示(美国专利us6512140b1)。
[0009][0010]
现有技术的制备方法多采用传统的反应釜,所用时间长达数小时甚至数十小时,会使反应物停留时间过长,导致副产物的产生。
技术实现要素:
[0011]
本发明的目的在于解决现有制备工艺存在的生产成本高、收率低、路线长、时间长等问题,提供一种孟鲁司特钠侧链中间体的制备工艺,以实现孟鲁司特钠侧链中间体的大规模生产。
[0012]
该制备工艺具体方案如下:
[0013]
本发明提供一种式1结构化合物的制备方法,技术路线如下:
[0014][0015]
具体地,技术路线与操作步骤如下:
[0016]
步骤(1):
[0017][0018]
制备物料a:将水、甲醇、1,1-环丙基二羧酸混合;
[0019]
制备物料b:将氢气加入甲醇/水混合物中;
[0020]
将物料a、b一起输送至固定床反应器中,在ru-sn-pt/c催化剂,或选用铜-铬催化剂,在1-10mpa,120~240℃条件下反应10~150s,换热介质为乙二醇或水和乙二醇的混合物;
[0021]
反应完成后的物料将溶剂减压脱溶至干,得化合物5。
[0022]
进一步的,催化剂选用采用添加sn和或pt、re等的负载型ru系催化剂,优选6%ru-5%sn-3%pt/c催化剂,或选用铜-铬催化剂。
[0023]
进一步的,反应温度为100℃~240℃,可以但不局限于100℃、120℃、160℃、200℃、240℃,优选为160℃。
[0024]
进一步的,反应压力为1mpa~10mpa,可以但不局限于1mpa、3mpa、5mpa、8mpa、10mpa,优选为6mpa。
[0025]
步骤(2):
[0026][0027]
在路易斯酸催化下,将化合物5与氯化氢气体混合物,得到化合物4。
[0028]
进一步的,1,1-环丙烷二甲醇、氯化氢气体的投料摩尔比为1:0.1~2.5,可以但不局限于1:0.1、1:0.2、1:0.5、1:0.8、1:1、1:1.2、1:1.5、1:1.7、1:2、1:2.3、1:2.5,优选为1:2.2。
[0029]
进一步的,催化剂是路易斯酸,包含但不限于氯化锌,三氯化铝,氯化镁。
[0030]
步骤(3):
[0031][0032]
将化合物4与氰化钠(氰化钾或其他盐)固体或水溶液加入有机溶剂中,搅拌反应得到化合物3。
[0033]
进一步的,化合物4、氰化物的投料摩尔比为1:0.1~2.5,优选为1:1.2,溶剂包括但不限于n,n-二甲基甲酰胺,乙腈,甲醇,水;氰化试剂是氰化钠、氰化钾、氢氰酸其中的一种,优选为氰化钠;反应温度为60~150℃,优选为90℃。
[0034]
步骤(4):
[0035][0036]
将化合物3与巯基试剂加入有机溶剂中,搅拌反应得到化合物2。
[0037]
进一步的,化合物3、巯基试剂的投料摩尔比为1:0.1~2.5,优选为1:1.1,溶剂包括但不限于n,n-二甲基甲酰胺,乙腈,甲醇;巯基试剂是硫脲,硫代乙酸钾,甲基磺酰氯其中的一种,优选为硫代乙酸钾;反应温度为30~90℃,优选为45℃。
[0038]
进一步的,反应温度为30~90℃,可以但不局限于30℃、45℃、60℃、75℃、80℃、85℃、90℃,优选为45℃。
[0039]
步骤(5):
[0040][0041]
将化合物2与碱加入溶剂中,搅拌反应得到化合物1。
[0042]
进一步的,化合物2、碱的投料摩尔比为1:0.1~2.5,优选为1:1.1,碱试剂是氢氧化钠,氢氧化钾其中的一种,优选为氢氧化钠;反应温度为30~80℃,优选为45℃。
[0043]
通过采用本发明的技术方案,达到的技术效果如下:
[0044]
1、本发明第一步反应采用连续固定床反应器,反应时间缩短至几百秒,安全性较传统反应釜有了质的提高,极大缩短了反应时间,增加了安全性;
[0045]
2、本发明步骤短,副产物少,环境友好,避免使用氯化亚砜,溴素等污染严重的试剂。
[0046]
3、本发明所述制备工艺路线短,简化了制备工艺步骤,比传统路线短2-3步;
[0047]
4、本发明采用连续化固定床反应器制备产物,可连续化生产,制备方法自动化程度高,方便控制,减少人力成本,有良好的工业化应用前景。
具体实施方式
[0048]
为了更好地理解本发明的内容,下面结合具体实施例来对本发明的技术方案进行清楚、完整地描述,但具体的实施方式并不是对本发明内容所作的限制。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0049]
实施例1
[0050][0051]
其中,
[0052][0053]
制备物料a:将甲醇(15l)、1,1-环丙烷二甲醇(100mol,7.6kg)、水(15l)混合,搅拌溶清,备用;
[0054]
制备物料b:氢气;
[0055]
通过柱塞泵分别将物料a、b输送至固定床反应器中,通过计数泵设置物料a、b的流速,使1,1-环丙烷二甲醇、氢气的摩尔配比为1:4,压力为6mpa,在160℃下保持反应进行10~150s;
[0056]
将反应液减压脱溶至干,干燥得5.94kg化合物5,收率99.7%。
[0057]
实施例2
[0058][0059]
将59.4g化合物5,1.0g氯化锌,46.7g氯化氢,乙腈300ml,混合物均匀,45℃反应4h,减压蒸干溶剂,得80.4g化合物4。
[0060]
实施例3
[0061][0062]
在反应瓶中加入化合物4(80.4g,582.6mmol),氰化钠(31.4g,640.9mmol),乙醇(550ml),缓慢升温至90℃,保温反应20小时。反应结束后降温至45℃,减压蒸除乙醇,往体系中加入甲苯200ml,水200ml,分液,甲苯相蒸干得到目标产物,纯度99.7%。
[0063]
实施例4
[0064]
[0065]
在反应瓶中加入化合物3(23.6g,182.6mmol),硫代乙酸钾(25.0g,219.1mmol),乙醇(200ml),缓慢升温至45℃,保温反应20小时。反应结束后减压蒸除乙醇,往体系中加入甲苯200ml,水200ml,分液,甲苯相蒸干得到目标产物2,收率94.3%,纯度98.7%。
[0066]
实施例5
[0067][0068]
在反应瓶中加入化合物2(13.9g,82.6mmol),氢氧化钠(3.6g,90.9mmol),乙醇(200ml),缓慢升温至45℃,保温反应20小时。反应结束后,减压蒸除乙醇,往体系中加入甲苯100ml,水100ml,分液。水相调ph=3-4,水相中加入甲苯100ml,甲苯相蒸干得到目标产物1,收率96.3%,纯度98.5%。
[0069]
以上所述,仅为本发明的较佳实施例而已,并非用于限定本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1