一种4-羟基-1,5-萘啶类铕配合物的生产制备方法与流程
1.本发明属于稀土配合物发光材料制备领域,具体涉及一种4-羟基-1,5
‑ꢀ
萘啶类铕配合物的生产制备方法。
背景技术:
2.稀土发光材料具有高效、色纯等特点,长期以来一直是国家科技开发的重点方向之一,在照明、显示等领域具有数百亿元/年的庞大市场。稀土发光材料根据组成可分为无机发光材料和有机配合物发光材料两类。与无机荧光粉相比,稀土配合物拥有更大的摩尔吸光系数,可以在低浓度或很少用量下表现出非常高的发光亮度。另外,稀土配合物还具有良好的相容性,可以掺入高分子中制备高透明度的发光薄膜,具有无机发光材料不可比拟的优势。
3.但在实际应用中,传统的稀土配合物发光材料却面临着光稳定性差的缺陷,极大地限制了其大规模应用。例如传统的β-二酮类稀土配合物在紫外线激发下很容易发生光降解现象,由于有机配体被迅速破坏,导致其发光强度迅速降低(synth.met.2011,161,964)。
4.在以往研究中,本研发团队开发出一种具有高发光效率和出色光稳定性的4-羟基-1,5-萘啶(简称萘啶)类稀土发光材料。这类材料不仅具有刚性的分子结构,而且可以形成紧密堆积来限制分子振动,使其抵抗光降解的能力大大提升。这种萘啶类稀土配合物在荧光防伪、转光农膜、照明、显示等领域都具有重要应用前景。
5.在产业化的过程中,本研发团队从实验室小试开始,开发了萘啶类稀土配合物的规模化生产工艺,并成功打通了这类配体和配合物的合成路线 (cn201110139842.1;adv.funct.mater.2016,26,2085)。
6.但在规模化生产上,原有合成路线还存在一些技术难点。
7.萘啶稀土配合物的合成方面,原有生产工艺是将配体与naoh在醇类溶剂中混合去掉质子,再与氯化稀土盐配位反应得到稀土配合物,由于反应生成的副产物nacl在醇中也会析出,存在nacl难以和稀土配合物产品分开的难题。对此,一种办法是蒸干醇类溶剂后,用大量其他良性溶剂(例如丙酮、二氯甲烷)去溶解配合物,然后将不溶的nacl滤除(adv. funct.mater.2016,26,2085),这样的话生产工艺复杂且成本较高。另一种方法是通过水洗的方式去除nacl(cn201110139842.1),该方法虽然操作简单,但发明人进行了大量实验研究发现,水分子活性和配位性都很强,大量水的引入容易对配合物产生一定破坏作用。
8.对于水洗除盐的方式,发明人进行了大量实验研究,发现存在两个弊端。一方面,水分子具有很强的配位性,会与其他中性配体进行竞争配位,从而造成稀土配合物发光被水分子的高频振动淬灭;另一方面,由于4
‑ꢀ
羟基-1,5-萘啶类配体是弱酸,所以与水分子接触容易导致阴离子配体水解和解离,并造成配位不饱和以及羟基配位桥联(水分子竞争配位以及配体水解和解离过程结构变化如下式所示)。
[0009][0010]
水分子竞争使中性配体离去
[0011][0012]
水分子使萘啶类配体水解并解离,且易产生羟基桥联
[0013]
因此,如何能够高效地去除副产物盐类、提高产物发光效率仍然存在技术难题。解决这些技术难点,提高生产效率,对该类材料的产业化生产和大规模市场应用非常重要。
技术实现要素:
[0014]
本发明的目的是为了解决萘啶类稀土配合物原有的制备工艺中存在的工艺复杂、产物发光效率低的技术难题,提供一种4-羟基-1,5-萘啶类铕配合物生产制备方法,可以有效提升产品发光效率、提高操作便捷性、降低生产成本。
[0015]
为实现上述技术目的,本发明的技术方案是:一种4-羟基-1,5-萘啶类铕配合物的生产制备方法,在醇类溶剂中,将4-羟基-1,5-萘啶类配体、有机季铵碱、三价铕盐、中性配体按摩尔量3:(3~3.3):1:(1~3)的比例混合,流温10~240分钟,冷却后过滤或离心分离得到所述4-羟基-1,5-萘啶类铕配合物;
[0016]
所述三价铕盐为三价铕的盐酸盐、硝酸盐或醋酸盐;
[0017]
所述4-羟基-1,5-萘啶类铕配合物的结构如式i所示:
[0018][0019]
式i中,r1、r2、r3、r4、r5选自氢原子、卤素原子、氰基、甲基、乙基、三氟甲基、五氟乙基中的任意一种;l为中性配体,x=1~3;
[0020]
所述中性配体为含氮芳香杂环类配体及其甲基或苯基取代的衍生物、含氮芳香杂环-n-氧化物类配体及其甲基或苯基取代的衍生物、甲基或苯基取代的膦酰类配体[(r)3po]。
[0021]
所述含氮芳香杂环类配体选自吡啶、咪唑、吡唑、噻唑、噁唑、噁二唑、三氮唑衍生出的单齿或多齿配体,更优选为上述基团连接组成的双齿或三齿配体。当所述含氮芳香杂环类配体选自上述结构时,化合物中的杂环氮原子拥有较高的电子云密度,因此具有较强的配位能力。所述含氮芳香杂环类配体举例为1,10-邻菲罗啉、2,2
′‑
联吡啶、2,2':6',2
”‑
三联吡啶、苯并咪唑、苯并噁唑、苯并噻唑、喹啉、异喹啉、2-(1-甲基咪唑-2-基)吡啶、 2-(吡唑-1-基)吡啶、2-(1,2,3三氮唑-1-基)吡啶、2-(吡啶-2-基)噁唑、2-(吡啶-2-基)噻唑、2-(吡啶-2-基)-1,3,4-噁二唑。
[0022]
所述含氮芳香杂环-n-氧化物类配体是指氮杂原子通过n-氧化得到的n-氧化物,其中的n-o键极性很大,o原子电子云密度高,具有较强的配位能力。所述含氮芳香杂环-n-氧化物举例为吡啶-n-氧化物、咪唑-n
‑ꢀ
氧化物、吡唑-n-氧化物、2,2
′‑
联吡啶-n-氧化物、1,10-邻菲罗啉-n-氧化物。
[0023]
上述铕配合物的生产制备方法中,特别优选的,所述中性配体选自1,10-邻菲罗啉(phen)及其甲基或苯基取代的衍生物、2,2
′‑
联吡啶(bpy) 及其甲基或苯基取代的衍生物、吡啶氮氧化物(pyno)、三苯基氧磷 (tppo)。
[0024]
上述部分中性配体的结构如式ii所示:
[0025][0026]
所述中性配体选择为具有较好堆积性的中性配体,所形成的铕配合物也具备较好堆积性,从而能够在醇类溶剂中析出。
[0027]
所述中性配体可以有很多变化和衍生化,本发明不限定具体的中性配体结构(只要具有良好堆积性),采用各种衍生的中性配体制备的铕配合物都在本专利的保护范围内。
[0028]
上述铕配合物的生产制备方法中,优选的,式i中,r
1-r5选自氢原子、f、cl、br、氰
基、甲基、乙基中的任意一种;进一步优选的,r
1-r5选自氢原子、cl、氰基、甲基中的任意一种。当r
1-r5选择上述基团时,所制备的4-羟基-1,5-萘啶类铕配合物具备较好的堆积性,从而能够在醇类溶剂中析出。r1、r2、r3、r4、r5可以相同,也可以不同。
[0029]
上述铕配合物的生产制备方法中,所述有机季铵碱优选为c1-c18的直链或支链烷基、苯基、苄基取代的氢氧化铵,进一步优选为四甲基氢氧化铵、四乙基氢氧化铵、四丁基氢氧化铵、苯基三甲基氢氧化铵、苄基三甲基氢氧化铵、十六烷基三甲基氢氧化铵中的任意一种。
[0030]
有机季铵碱相对于4-羟基-1,5萘啶配体的比例可以等当量或稍微过量(1~1.1当量),但不能过量太多,否则羟基也会参与竞争配位,导致不能生成理想比例的配合物。
[0031]
上述铕配合物的生产制备方法中,所述醇类溶剂选自甲醇、乙醇或者二者的混合溶剂。
[0032]
上述铕配合物的生产制备方法中,醇类溶剂的量是4-羟基-1,5-萘啶类配体质量的5~50倍。
[0033]
本发明的有益效果在于:由于采用了有机季铵碱去除4-羟基-1,5-萘啶类配体的质子,与稀土离子配位后,生成的有机铵盐可以溶于醇类溶剂中,而稀土配合物在醇类溶剂中直接析出,可以通过过滤或离心分离得到较纯的稀土配合物产品。本发明制备方法简单易操作,且不会发生由于水洗而导致的竞争配位或萘啶类配体水解的问题,产品能够保持高的发光效率,效果突出。
[0034]
需要特别说明的是,本发明的技术方案并不采用有机胺、吡啶等有机弱碱来制备铕配合物(虽然副产物有机胺盐、吡啶盐也能够溶于醇类溶剂,方便去除)。原因是,经发明人研究发现,4-羟基-1,5-萘啶类配体是弱酸 (测试pka约为10),需要强碱才能使其充分去质子化。对比实验发现,若采用有机弱碱(如三乙胺、吡啶等)制备稀土配合物,萘啶类配体并不能够完全去质子化,导致配合物产品中存在酮式结构配位的分子(示意如下式所示,其中的nh键振动会带来淬灭),或者未去质子化的配体不能参与配位从而造成配体不足,发光效率不高。
[0035][0036]
本发明所用的有机季铵碱为强碱,可以使4-羟基-1,5萘啶类配体充分去质子化,从而可以得到高发光效率的铕配合物产品。
附图说明
[0037]
图1是本发明实施例1所制备的4-羟基-1,5-萘啶铕配合物的光致发光量子效率测试曲线。
[0038]
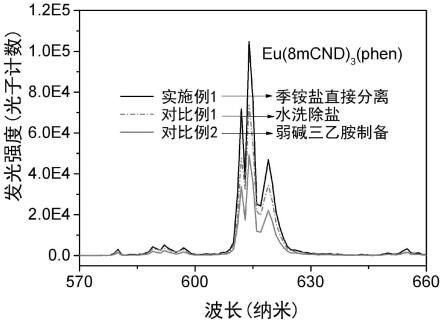
图2是本发明实施例1所制备4-羟基-1,5-萘啶铕配合物与对比例1 采用水洗方法去除氯化钠以及对比例2用弱碱三乙胺所制备的铕配合物的发光强度对比图(激发波长为
350nm)。
具体实施方式
[0039]
下面通过具体实施例对本发明的产品、制备方法作进一步的说明,但这些具体实施方案不以任何方式限制本发明的保护范围。
[0040]
在醇类溶剂中,按摩尔量将4-羟基-1,5-萘啶类配体(3份)、有机季铵碱(3~3.3份)混合去除配体的质子,然后加入三氯化铕(1份)、中性配体(1~3份),回流10~240分钟,冷却后直接过滤或离心分离即可得到所述4-羟基-1,5-萘啶类铕配合物。
[0041]
《铕配合物合成》
[0042]
实施例1.
[0043]
铕配合物eu(8mcnd)3(phen)合成,本实施例铕配合物结构如下所示:
[0044][0045]
在800ml无水乙醇中,加入配体3-氰基-4-羟基-8甲基-1,5-萘啶 (h8mcnd)55.5g(0.3mol)、有机碱-五水合四甲基氢氧化铵54.3g (0.3mol)、六水合三氯化铕36.6g(0.1mol)、无水邻菲罗啉18.0g(0.1mol),加热回流2小时。生成白色铕配合物固体,而副产物-四甲基氯化铵溶于乙醇。冷却后,直接过滤分离,乙醇洗涤,真空烘干,得到白色铕配合物产品81.4g,收率92%。质谱分析(m/z,esi):计算值885.1,实验值886.1 [m+h]
+
。元素分析(质量百分含量%):c,55.96(55.88);h,3.23(3.13),n,16.95(17.07),括号中为eu(8mcnd)3(phen)
·
1.0h2o理论值。
[0046]
实施例2.
[0047]
铕配合物eu(8mcnd)3(tppo)2合成,制备方法同实施例1,只是将中性配体换为三苯基氧化膦(tppo),有机碱换为四丁基氢氧化铵。本实施例铕配合物结构如下所示:
[0048]
[0049]
在800ml无水乙醇中,加入配体3-氰基-4-羟基-8甲基-1,5-萘啶 (h8mcnd)55.5g(0.3mol)、有机碱-四丁基氢氧化铵(2m乙醇溶液) 150ml(0.3mol)、六水合三氯化铕36.6g(0.1mol)、三苯基氧化膦(tppo) 55.7g(0.2mol),加热回流2小时。生成白色铕配合物固体,而副产物
‑ꢀ
四丁基氯化铵溶于乙醇。冷却后,直接过滤分离,乙醇洗涤,真空烘干,得到白色铕配合物产品112.2g,收率89%。质谱分析(m/z,esi):计算值1261.2,实验值1262.2[m+h]
+
。元素分析(质量百分含量%):c,62.07 (62.13);h,4.30(4.15);n,9.85(9.65),括号中为eu(8mcnd)3(tppo)2·
0.8 c2h5oh
·
0.5h2o理论值。
[0050]
实施例3.
[0051]
铕配合物eu(nd)3(pyno)3合成,制备方法同实施例1,只是配体换为4-羟基-1,5萘啶(hnd),中性配体换为吡啶-n-氧化物(pyno),溶剂换为甲醇。本实施例铕配合物结构如下所示:
[0052][0053]
在50ml甲醇中,加入配体4-羟基-1,5萘啶(hnd)4.38g(0.03mol)、有机碱-五水合四甲基氢氧化铵5.43g(0.03mol)、六水合三氯化铕3.66g (0.01mol)、吡啶-n-氧化物2.85g(0.03mol),加热回流2小时。生成白色铕配合物固体,而副产物-四甲基氯化铵溶于甲醇。冷却后,直接过滤分离,甲醇洗涤,真空烘干,得到白色铕配合物产品7.42g,收率85%。质谱分析(m/z,esi):计算值873.1,实验值874.1[m+h]
+
。元素分析(质量百分含量%):c,52.33(52.27);h,3.87(3.90);n,13.76(13.72),括号中为eu(nd)3(pyno)3·
1.0ch3oh
·
0.8h2o理论值。
[0054]
对比例1.
[0055]
铕配合物eu(8mcnd)3(phen)合成,制备方法同实施例1,只是将碱换为无机碱氢氧化钠,后处理方法采用水洗方式去除nacl。本实施例铕配合物的目标结构如式iii所示(实际含水解和桥联复杂结构)。
[0056]
在800ml无水乙醇中,加入配体8mcnd 55.5g(0.3mol)、无机碱
‑ꢀ
氢氧化钠12.0g(0.3mol)、六水合三氯化铕36.6g(0.1mol)、无水邻菲罗啉18.0g(0.1mol),加热回流2小时。生成白色铕配合物沉淀,以及氯化钠沉淀。冷却后,过滤分离产品,用200ml水洗涤除去nacl,真空烘干,得到白色铕配合物产品72.5g,收率82%。质谱分析(m/z,esi):计算值885.1,实验值886.1[m+h]
+
。
[0057]
对比例2.
[0058]
铕配合物eu(8mcnd)3(phen)合成,制备方法同实施例1,只是将碱换为有机弱碱-三乙胺。本实施例铕配合物的目标结构如式iii所示(实际含未去质子化的酮式配位结构)。
[0059]
在800ml无水乙醇中,加入配体3-氰基-4-羟基-8甲基-1,5-萘啶 (h8mcnd)55.5g(0.3mol)、有机碱-三乙胺30.3g(0.3mol)、六水合三氯化铕36.6g(0.1mol)、无水邻菲罗啉18.0g(0.1mol),加热回流 2小时。生成白色铕配合物固体,而副产物-三乙胺盐酸盐溶于乙醇。冷却后,直接过滤分离,乙醇洗涤,真空烘干,得到白色铕配合物产品78.7g,收率89%。质谱分析(m/z,esi):计算值885.1,实验值886.1[m+h]
+
。
[0060]
《铕配合物表征》
[0061]
实施例6.
[0062]
(1)激发与发射光谱
[0063]
激发、发射光谱是采用爱丁堡fls 1000荧光光谱仪测试的,激发光源为氙灯,检测器为光电倍增管。石英样品池中放置适量铕配合物粉末,激发与发射狭缝均选择0.5nm。在350nm紫外光激发波长下测试发射光谱。
[0064]
(2)光致发光量子效率
[0065]
光致发光量子效率是通过fls 1000光谱仪的积分球测得的绝对量子效率。以350nm为激发波长,以空白石英池为参比扫描340~750nm范围的背景信号,石英样品池中放置适量铕配合物粉末扫描样品的吸收及发光信号;对两次扫描的谱图进行处理,340~360nm范围的光谱积分峰面积的差值为激发光的吸收值,500~750nm范围的光谱积分峰面积的差值为发光强度值;发光强度值与吸收值之间的比值即为光致发光量子效率。
[0066]
(3)溶解性测试
[0067]
取50mg铕配合物样品,在2ml塑料离心管中,加入1ml良溶剂dmf (n,n-二甲基甲酰胺),超声1分钟,观测溶解情况。
[0068]
测试结果分析:
[0069]
测试结果如图1、2及表1所示。图1是实施例1所制备的铕配合物的光致发光量子效率测试曲线,实施例1使用有机强碱四甲基氢氧化铵制备的铕配合物的光致发光量子效率为62%。图2是实施例1与对比例1、对比例2所制备的铕配合物的发光强度对比图,激发波长为350nm。实施例1使用有机强碱四甲基氢氧化铵制备4-羟基-1,5-萘啶铕配合物,对比例 1采用水洗方法去除氯化钠,对比例2用弱碱三乙胺制备铕配合物,实施例1产物的发光强度大大高于对比例1和对比例2产物的发光强度。
[0070]
从表1可见,基于8mcd配体,实施例1使用有机强碱四甲基氢氧化铵制备的铕配合物的光致发光量子效率为62%;而对比例1中,采用氢氧化钠作碱,通过水洗方法去除氯化钠,由于出现配体水解和解离,所制备的铕配合物的光致发光量子效率仅为44%;对比例2使用有机弱碱三乙胺制备铕配合物,由于三乙胺碱性较弱,不能使萘啶类配体充分去质子化,产品中存在大量酮式结构配位的分子(nh键造成振动淬灭),以及大量配位不饱和的eu(8mcnd)2cl(phen)分子,光致发光量子效率仅为29%。
[0071]
溶解性方面,对比例1中,大量水洗除盐的方式使萘啶类配体发生水解和解离,得到的铕配合物在良溶剂丙酮/dmf(n,n-二甲基甲酰胺)中不能全溶,有较多沉淀,证明产生一定形式的复杂桥联结构。对比例2 采用弱碱三乙胺制备铕配合物,由于产品中存在大量配位不饱和的 eu(8mcnd)2cl(phen)分子,引起复杂桥联,所以在dmf中也不能全溶。
[0072]
表1铕配合物发光性质表征
[0073][0074]
由于本发明采用有机季铵碱这样的有机强碱去除4-羟基-1,5-萘啶类配体的质子并与铕离子反应制备铕配合物,能使4-羟基-1,5-萘啶类铕配体充分去质子化,且反应得到的铵盐溶于乙醇中,生成的稀土配合物可在乙醇中直接析出,使制备和分离方法简单便捷,并使铕配合物产品保持高的发光效率。本发明制备方法简单易操作,且不会发生由于水洗而导致的竞争配位或萘啶类配体水解的问题,产品能够保持高的发光效率,效果突出。
[0075]
以上所述实施例仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1