一种可变形双重响应性高载药量聚前药纳米载体的制备方法
1.本发明涉及药物制剂领域,具体涉及一种可变形双重响应性高载药量聚前药纳米载体的制备方法。
背景技术:
2.众所周知,癌症在生物医学领域又被称为恶性肿瘤,是正常的细胞发生突变的结果。其生物学特征为快速增殖,通过逃避生长抑制剂对细胞死亡的抵抗力,从而实现复制性永生,侵袭和转移。由于癌症极易转移,现在的医学水平很少做到完全治愈癌症,使得癌症已成为对全球医疗保健领域的巨大挑战。
3.喜树碱()是一类非常著名的抗癌药物,由于其药效显著,因而广受人们的喜爱。其分子式为,从临床实验到应用一共经历了十年的时间。后来广大研究者们又在前人的基础上进行了数年的系统研究,现已成功研制出了多种喜树碱衍生物类抗癌药物。研究表明细胞核中的拓扑异构酶有掌控和影响细胞代谢时的拓扑状态的能力,其通过相应的生理作用使不能复制,进而造成不可逆的链破坏,导致细胞凋亡。虽然喜树碱具有显著的治疗效果,但是作为一种小分子药物使得它也存在种种缺点。为了提高的生物利用度和溶解度,经常使用改性过的单体药物通过相应的聚合方法得到特定的两亲性聚合物,进而有效的提高了其溶解度和载药量。
4.为解决传统化学药物存在的不足,研究人员经过大胆的尝试,并在前人的基础上发展出了纳米药物递送系统。纳米医学为纳米科学的一个在医学领域的分支,也属于一个集合了众多领域的交叉领域。近年来有着多种功能的纳米药物在克服了重重困难之后,终于将治疗肿瘤的相关试剂送至肿瘤部位。
5.所谓的前药即是通过化学键把相关药物连接到分子基体上,此时药物不具备活性,只有通过相应的生物体内的转化,才能使化学键断裂释放出活性药物,进而发挥作用。为了解决像载药量和载药率都较低、胶束稳定性较差、还存在过早释放的缺点等相关问题,科学家们提出了一种解决方案,通过化学键将药物连接在大分子上。即提出了前药的概念。相对于聚合物胶束,前药大大提高了药物的载药率和载药量,也改善了其稳定性,具备很大的优势。
6.为了延长纳米载体的血液循环时间,从而使其到达特定的肿瘤部位发挥治疗效果,研究人员进行了大量的相关工作。研究人员发现在纳米载体表面加一层亲水性的聚乙二醇,由于乙二醇带有微弱的负电荷,进而延长了纳米载体在血液循环中的时间,进而改善了纳米载体的治疗效果。但是尽管纳米载体的聚乙二醇化可以让其在血液循环中达到较好的“隐身”效果,而被广泛应用,但是聚乙二醇化也将增大纳米载体尺寸,限制了纳米载体表面基团与生物界面的相互作用,进而阻碍它们的组织渗透能力和与细胞的结合能力,大大影响了纳米载体的抗肿瘤功效。
7.众所周知,刺激响应性前药在降低药物毒性进而降低药物对机体的毒副作用方面很安全有效,但是影响其成为一种高效的药物的主要限制因素是刺激响应性前药自身比较
低的药效。而为了达到治疗效果,使得需要不可避免地引入大量的药物载体以提高药效,但是多次给药又有导致肿瘤细胞产生耐药性的后果。因此如何提高前药的载药量以解决实际临床应用时药效不足的难题,是该领域需要急切解决的挑战。
技术实现要素:
8.本发明的目的在于提供一种可变形双重响应性高载药量聚前药纳米载体的制备方法,其大大提高了喜树碱药物的载药量,双重响应性的设计不仅可以实现药物的可控释放,还可以通过酸响应性去除聚乙二醇链使得纳米载体的尺寸变小,进而提高了纳米载体与细胞的结合能力,提高抗肿瘤功效。
9.为实现上述目的,本发明提供如下技术方案:
10.一种可变形双重响应性高载药量聚前药纳米载体的制备方法,所述方法包括以下步骤:
11.(1)三聚体结构的喜树碱前药单体的制备:以均三苄醇为起始原料,通过保护羟基,硝化带上硝基,氢化变成氨基,三光气活化氨基后通过反应带上甲基丙烯酸酯类的二硫化物,酸性离子交换树脂脱保护,连接上喜树碱药物分子,形成了喜树碱前药单体;
12.(2)带双键的酸响应性可去除聚乙二醇的制备:以分子量2000的甲氧基聚乙二醇为起始原料,通过对甲苯磺酰氯活化羟基进而通过与叠氮化钠反应带上叠氮基团,把叠氮基团氢化成氨基,得到氨基聚乙二醇,氨基聚乙二醇与炔基官能化的对羟基苯甲醛反应得到酸响应性聚乙二醇,与开环后的甲基丙烯酸缩水甘油酯反应,得到带双键的酸响应性可去除聚乙二醇;
13.(3)双重响应性高载药量聚前药的制备:由基于三聚体结构的谷胱甘肽响应性喜树碱前药单体和带双键的酸响应性可去除的聚乙二醇通过可逆加成-断裂链转移聚合,形成了双重响应性高载药量聚前药;
14.(4)可变形双重响应性高载药量聚前药纳米载体的制备:将制备的双重响应性高载药量聚前药在去离子水中自组装从而得到可变形双重响应性高载药量聚前药纳米载体。
15.优选地,所述喜树碱前药单体的制备方法具体包括以下步骤:
16.a、带端羟基和二硫键的甲基丙烯酸酯类分子的合成:
17.将甲基丙烯酰氯和双羟乙基二硫化物反应,分离,洗涤并干燥后得到带端羟基和二硫键的甲基丙烯酸酯类分子;
18.b、叔丁基二甲基氯硅烷保护均三苄醇的三个-oh产物的合成:
19.将均三苄醇和叔丁基二甲基氯硅烷反应,分离,洗涤并干燥后得到叔丁基二甲基氯硅烷保护均三苄醇的三个-oh产物;
20.c、已保护均三苄醇-oh的硝化产物的合成:
21.将保护均三苄醇的三个-oh产物和浓硝酸反应,分离,洗涤并干燥后得到已保护均三苄醇-oh的硝化产物;
22.d、已保护均三苄醇-oh的氢化产物的合成:
23.将已保护均三苄醇-oh的硝化产物和水合肼进行氢化反应,分离,洗涤并干燥后得到已保护均三苄醇-oh的氢化产物;
24.e、含二硫键和已保护均三苄醇-oh的甲基丙烯酸酯类分子的合成:
25.将已保护均三苄醇-oh的氢化产物和三光气反应,分离,洗涤并干燥后得到带端-nco和已保护均三苄醇-oh产物;
26.将已保护均三苄醇-oh产物和带端-nco产物和带端羟基和二硫键的甲基丙烯酸酯类分子反应,分离,洗涤并干燥后得到含二硫键和已保护均三苄醇-oh的甲基丙烯酸酯类分子;
27.f、含二硫键和均三苄醇-oh脱保护的甲基丙烯酸酯类分子的合成:
28.将已保护均三苄醇-oh的甲基丙烯酸酯类分子和酸性离子交换树脂反应,分离,洗涤并干燥后得到含二硫键和均三苄醇-oh脱保护的甲基丙烯酸酯类分子;
29.g、三聚体结构的喜树碱前药单体的合成:
30.将含二硫键和均三苄醇-oh脱保护的甲基丙烯酸酯类分子和喜树碱经三光气反应后的酰氯中间体反应,分离,洗涤并干燥后得到三聚体前药结构的喜树碱前药单体。
31.优选地,所述步骤b中,均三苄醇、叔丁基二甲基氯硅烷的摩尔比为1:4.5。
32.优选地,所述步骤c中,保护均三苄醇的三个-oh产物和浓硝酸反应在零下20℃的环境下进行。
33.优选地,所述步骤d中,氢化反应在钯碳的催化作用下进行。
34.优选地,所述带双键的酸响应性可去除聚乙二醇的制备方法具体包括以下步骤:
35.a、对甲苯磺酰氯官能化聚乙二醇的合成:
36.将对甲苯磺酰氯和甲氧基聚乙二醇反应,洗涤,在冷乙醚中沉淀,分离并干燥后得到对甲苯磺酰氯官能化聚乙二醇;
37.b、叠氮化物官能化聚乙二醇的合成:
38.将叠氮化钠和对甲苯磺酰氯官能化聚乙二醇反应,分离,洗涤并干燥后得到叠氮化物官能化聚乙二醇;
39.c、氨基官能化聚乙二醇的合成:
40.将水合肼和叠氮化物官能化聚乙二醇反应,分离,洗涤并干燥后得到氨基官能化聚乙二醇;
41.d、炔基官能化的对羟基苯甲醛的合成:
42.将溴丙炔和对羟基苯甲醛反应,分离,洗涤并干燥后得到炔基官能化的对羟基苯甲醛;
43.e、炔基官能化的酸响应性聚乙二醇的合成:
44.将炔基官能化的对羟基苯甲醛和氨基官能化聚乙二醇反应,分离,洗涤并干燥后得到炔基官能化的酸响应性聚乙二醇;
45.f、叠氮官能化的甲基丙烯酸缩水甘油酯的合成:
46.将甲基丙烯酸缩水甘油酯和叠氮化钠反应,分离,洗涤并干燥后得到叠氮官能化的甲基丙烯酸缩水甘油酯;
47.g、带双键的酸响应性可去除聚乙二醇的合成:
48.将叠氮官能化的甲基丙烯酸缩水甘油酯和炔基官能化的酸响应性聚乙二醇反应,分离,洗涤并干燥后得到带双键的酸响应性可去除聚乙二醇。
49.优选地,所述双重响应性高载药量聚前药的合成方法具体如下:将带双键的酸响应性可去除聚乙二醇和三聚体结构的喜树碱前药单体通过可逆加成-断裂链转移聚合反
应,分离,洗涤并干燥后得到双重响应性高载药量聚前药。
50.优选地,所述步骤(3)中,一定要在无水无氧的环境下进行反应。
51.优选地,所述可变形双重响应性高载药量聚前药纳米载体的合成方法具体如下:将双重响应性高载药量聚前药溶于四氢呋喃中,将去离子水缓慢滴加到快速搅拌的四氢呋喃溶液中,滴加完毕后透析24小时,从而得到可变形双重响应性高载药量聚前药纳米载体。
52.一种所述的可变形双重响应性高载药量聚前药纳米载体的制备方法制备得到的可变形双重响应性高载药量聚前药纳米载体。
53.与现有技术相比,本发明的有益效果是:
54.1)本发明制备方法中的各步反应安全可靠,方便操作,各步转化率较高。
55.2)本发明中,三聚体结构的喜树碱前药单体在中间通过双羟乙基二硫化物引入了谷胱甘肽响应性断裂的二硫键,从而满足了前药单体的肿瘤特异性微环境的可控释放,提高了负载药物的稳定性,降低了细胞毒性;最后一步键合喜树碱药物分子时,苯环上的三个羟基均可键合喜树碱分子,得到三聚体喜树碱前药单体,当二硫键被谷胱甘肽切断时,通过分子重排,相继可以放出三个喜树碱药物分子,短时间内可以使肿瘤细胞内的药物浓度快速提高,弥补了传统前药单体载药量低的缺点;三聚体结构的喜树碱前药单体不仅大大提高了喜树碱药物的载药量,还可以用于稳定药物、输运和可控释放。
56.3)本发明中,酸响应性可去除的聚乙二醇链,在酸性的肿瘤微环境中,其改性后的聚乙二醇中的席夫碱键具有ph响应性以断裂去除聚乙二醇链,释放出由两亲性共聚物组装并包裹的喜树碱分子胶束,去除聚乙二醇链使得纳米载体尺寸变小,进而提高了纳米载体与细胞的结合能力,提高抗肿瘤功效。
57.4)本发明中,经组装后形成的纳米载体,胶束外层的亲水链为聚乙二醇修饰,无毒,具有良好的生物相容性,疏水内核由喜树碱分子经π-π堆叠而成,非常稳定,也提高了其抗肿瘤功效。
附图说明
58.图1为本发明实施例1中化合物1、化合物2、化合物3的核磁共振谱图;
59.图2为本发明实施例1中带端羟基和二硫键的甲基丙烯酸酯类分子的核磁共振谱图;
60.图3为本发明实施例1中化合物5和化合物6的核磁共振谱图;
61.图4为本发明实施例1中喜树碱前药单体的核磁共振谱图;
62.图5为本发明实施例2中对甲苯磺酰氯官能化聚乙二醇、叠氮化物官能化聚乙二醇、氨基官能化聚乙二醇的核磁共振谱图;
63.图6为本发明实施例2中炔基官能化的对羟基苯甲醛和炔基官能化的酸响应性聚乙二醇的核磁共振谱图;
64.图7为本发明实施例2中叠氮官能化的甲基丙烯酸缩水甘油酯的核磁共振谱图;
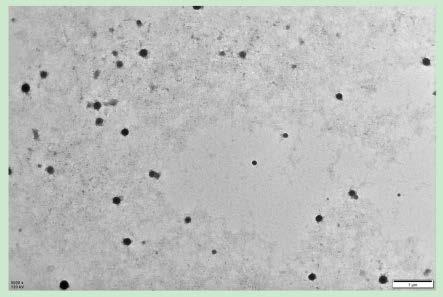
65.图8为本发明实施例2中带双键的酸响应性可去除聚乙二醇的核磁共振谱图;
66.图9为本发明实施例3中双重响应性高载药量聚前药的核磁共振谱图;
67.图10的(a)、(b)分别为本发明实施例3中双重响应性高载药量聚前药的紫外谱图和荧光谱图;
68.图11中(a)为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体的动态光散射图,(b)为可变形双重响应性高载药量聚前药纳米载体在ph:5.0的酸处理前后的zeta电位图;
69.图12为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在ph(5.0)处理2小时、以及在dtt(15mm)处理6小时和24小时后的动态光散射图;
70.图13为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体的透射电子显微镜图;
71.图14为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在ph(5.0)处理2小时后的透射电子显微镜图;
72.图15为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在dtt(15mm)处理24小时后的透射电子显微镜图;
73.图16(a)、(b)分别为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在dtt(15mm)作用下以及在dtt(15mm)和ph(5.0)的同时作用下,喜树碱原药分子释放曲线图。
具体实施方式
74.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
75.实施例1
76.三聚体结构的喜树碱前药单体的合成,具体包括以下步骤:
77.a、带端羟基和二硫键的甲基丙烯酸酯类分子的合成:
78.将双羟乙基二硫化物(3.24g,21mmol)和无水三乙胺(et3n 3.04g,30mmol)溶解在无水thf(四氢呋喃)中,将甲基丙烯酰氯(2.09g,20mmol)溶解在无水thf中,冰浴缓慢加入,反应液恢复至室温搅拌过夜,通过tlc板确定反应完成后,通过过滤,滤液浓缩,再用二氯甲烷重新溶解后,并用去离子水洗涤两次,饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,过硅胶柱提纯(洗脱剂为石油醚和乙酸乙酯,其中石油醚与乙酸乙酯的体积比为3:1),收集产物,真空干燥,得到带端羟基和二硫键的甲基丙烯酸酯类分子,其结构式为:
[0079][0080]
图2验证了本实施例中的带端羟基和二硫键的甲基丙烯酸酯类分子的成功制备。
[0081]
b、叔丁基二甲基氯硅烷保护均三苄醇的三个-oh产物(化合物1)的合成:
[0082]
将均三苄醇(1g,5.9mmol)溶解在无水dmf(n,n-二甲基甲酰胺)中,将叔丁基二甲基氯硅烷(tbscl 4.03g,26.7mmol)溶解在无水dmf中,冰浴缓慢加入,反应恢复至室温搅拌过夜,通过tlc板确定反应完成后,通过过滤,蒸发除去溶剂,用二氯甲烷重新溶解后,用去
离子水洗涤两次,饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,过硅胶柱提纯(洗脱剂为石油醚和乙酸乙酯,其中石油醚与乙酸乙酯的体积比为20:1),收集产物,真空干燥,得到产物1,其结构式为:
[0083][0084]
c、已保护均三苄醇-oh的硝化产物(化合物2)的合成:
[0085]
将乙酸酐(ac2o 15ml)冷却至5℃,逐滴加入浓硝酸(1.5ml,质量分数%)。加入完毕后在室温下搅拌15min,冷却至-20℃,化合物1(1.0g,1.9mmol)溶解在8ml乙酸酐溶液中,-20℃下缓慢加入。将反应恢复至0℃,搅拌60min。反应完成后,用乙酸乙酯稀释,用饱和碳酸氢钠溶液洗涤10次,至不在冒气泡,然后用饱和氯化钠溶液洗涤2次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,过硅胶柱提纯(洗脱剂为石油醚和乙酸乙酯,其中石油醚与乙酸乙酯的体积比为20:1),收集产物,真空干燥,得到产物2,其结构式为:
[0086][0087]
d、已保护均三苄醇-oh的氢化产物(化合物3)的合成:
[0088]
将化合物2(1.2g,2.3mmol)溶于无水甲醇溶液(meoh)中。在混合物中加入催化作用的钯碳(pd/c),然后缓慢加入水合肼(n2h44ml),反应混合物在65℃下加热回流搅拌过夜,通过tlc板确定反应完成后,通过过滤除去钯碳,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,用饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,过硅胶柱提纯(洗脱剂为石油醚和乙酸乙酯,其中石油醚与乙酸乙酯的体积比为20:1),收集产物,真空干燥,得到产物3,其结构式为:
[0089][0090]
图1验证了本实施例中的化合物1、化合物2、化合物3的成功制备。
[0091]
e、含二硫键和已保护均三苄醇-oh的甲基丙烯酸酯类分子(化合物5)的合成:
[0092]
将化合物3(0.4g,0.7mmol)和dmap(4-二甲氨基吡啶,0.22g,1.7mmol)溶解在无水二氯甲烷中,室温下逐滴加入三光气(0.074g,0.2mmol)的无水二氯甲烷溶液,室温下搅拌反应过夜,用tlc板确定反应完成后,得到带端-nco和已保护均三苄醇-oh产物,即为化合物5,化合物5的结构式为:
[0093][0094]
然后再加入3ml带端羟基和二硫键的甲基丙烯酸酯类分子(0.35g,0.7mmol)的无水二氯甲烷溶液,室温下搅拌反应12h,通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,用饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,过硅胶柱提纯(洗脱剂为石油醚和乙酸乙酯,其中石油醚与乙酸乙酯的体积比为5:1),收集产物,真空干燥,得到产物5,其结构式为:
[0095][0096]
f、含二硫键和均三苄醇-oh脱保护的甲基丙烯酸酯类分子(化合物6)的合成:
[0097]
将330mg化合物5和300mg amberlyst 15溶于14ml的dcm/meoh(二氯甲烷/甲醇8ml/6ml)混合溶剂中,室温下搅拌反应过夜,通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,过硅胶柱提纯(洗脱剂为无水甲醇和乙酸乙酯,其中无水甲醇和乙酸乙酯的体积比为1:15),收集产物,真空干燥,得到产物6,其结构式为:
[0098][0099]
图3验证了本实施例中的化合物5和化合物6的成功制备。
[0100]
g、三聚体前药结构的喜树碱前药单体(m-cpt-3)的合成:
[0101]
将喜树碱(0.347g,1.07mmol)和dmap(0.393g,3.22mmol)溶于无水二氯甲烷中,室温下逐滴加入三光气(0.129g,0.43mmol)的无水二氯甲烷溶液,室温下搅拌1h,用tlc板确定反应完成后,加入2ml的化合物6(0.14g,0.33mmol)的无水四氢呋喃溶液,室温下搅拌反应过夜,通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,过硅胶柱提纯(洗脱剂为无水甲醇和乙酸乙酯,其中无水甲醇和乙酸乙酯的体积比为1:30),收集产物,真空干燥,得到产物m-cpt-3,其结构式为:
[0102]
所述的喜树碱前药单体的合成路线为:
[0103][0104]
图4验证了本实施例中的三聚体结构的喜树碱前药单体的成功制备。
[0105]
实施例2
[0106]
带双键的酸响应性可去除聚乙二醇合成,所述方法包括以下步骤:
[0107]
a、对甲苯磺酰氯官能化聚乙二醇的合成:
[0108]
将2g甲氧基聚乙二醇和1.91g对甲苯磺酰氯溶解于dcm(20ml)中,加入1g无水三乙胺。室温下搅拌反应过夜。通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,用饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,然后从冷乙醚中沉淀,在室温下真空干燥,得到对甲苯磺酰氯官能化聚乙二醇,其结构式为:
[0109][0110]
b、叠氮化物官能化聚乙二醇的合成:
[0111]
将2g对甲苯磺酰氯官能化聚乙二醇和1g叠氮化钠溶解于dmf(10ml)中。室温下搅拌反应过夜,通过tlc板确定反应完成后,通过过滤,分离,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,用饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,然后从冷乙醚中沉淀,在室温下真空干燥,得到叠氮化物官能化聚乙二醇,其结构式为:
[0112][0113]
c、氨基官能化聚乙二醇的合成:
[0114]
将2g叠氮化物官能化聚乙二醇和0.2g还原催化剂钯碳溶解于dcm(20ml)中,置于零下20度的低温中搅拌15分钟,然后加入5ml水合肼,转至常温下搅拌24反应。通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,用饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,然后从冷乙醚中沉淀,在室温下真空干燥,得到氨基官能化聚乙二醇,其结构式为:
[0115][0116]
图5验证了本实施例中的对甲苯磺酰氯官能化聚乙二醇、叠氮化物官能化聚乙二醇、氨基官能化聚乙二醇的成功制备。
[0117]
d、炔基官能化的对羟基苯甲醛的合成:
[0118]
将12.22g对羟基苯甲醛和18.44g无水碳酸钾溶解于180ml无水丙酮(me2co)中,然后再加入用无水甲苯(60ml)溶解的溴丙炔,在70℃下回流反应12小时。通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用二氯甲烷重新溶解后,用饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,在室温下用无水乙醇溶解后,在零下20度的低温中重结晶,在室温下真空干燥,得到炔基官能化的对羟基苯甲醛,其结构式为:
[0119][0120]
e、炔基官能化的酸响应性聚乙二醇的合成:
[0121]
将2g氨基官能化聚乙二醇和0.1g无水碳酸钾,以及0.1g无水硫酸钠溶解于dcm(20ml)中,加入用5ml无水dcm溶解的炔基官能化的对羟基苯甲醛。室温下搅拌反应过夜。通过tlc板确定反应完成后,通过过滤,旋转蒸发除去溶剂,再用乙酸乙酯重新溶解后,用饱和
氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,然后从冷乙醚中沉淀,在室温下真空干燥,得到炔基官能化的酸响应性聚乙二醇,其结构式为:
[0122][0123]
图6验证了本实施例中的炔基官能化的对羟基苯甲醛和炔基官能化的酸响应性聚乙二醇的成功制备。
[0124]
f、叠氮官能化的gma(甲基丙烯酸缩水甘油酯)的合成:
[0125]
将11.3g的甲基丙烯酸缩水甘油酯、5.0g叠氮化钠、7.57g碳酸氢钠和0.16g的无水硫酸钠溶于120ml的thf/h2o(体积比为5/1)混合溶剂中,反应液升温至50℃搅拌反应过夜,通过tlc板确定反应完成后,通过过滤,滤液浓缩,再用二氯甲烷重新溶解后,并用去离子水洗涤两次,饱和氯化钠溶液洗涤两次,收集有机相,用无水硫酸钠干燥3h,抽滤除去无水硫酸钠,旋蒸除去溶剂,过硅胶柱提纯(洗脱剂为石油醚和乙酸乙酯,其中石油醚与乙酸乙酯的体积比为5:1),收集产物,真空干燥,得到叠氮官能化的gma,其结构式为:
[0126][0127]
图7验证了本实施例中的三聚体结构的叠氮官能化的甲基丙烯酸缩水甘油酯的成功制备。
[0128]
g、带双键的酸响应性可去除聚乙二醇的合成:
[0129]
将0.4g炔基官能化的酸响应性聚乙二醇和0.07g叠氮官能化的gma以及0.05gpmdeta(五甲基二乙烯三胺)溶解于4ml的dmf中,在氮气保护下加入加入0.03g溴化亚铜。在室温下搅拌反应过夜。通过tlc板确定反应完成后,通过过硅胶柱除去铜盐,旋转蒸发除去溶剂,再用二氯甲烷重新溶解后,从冷乙醚中沉淀,在室温下真空干燥,得到带双键的酸响应性可去除聚乙二醇,其结构式为:
[0130][0131]
所述的带双键的酸响应性可去除聚乙二醇的合成路线为:
[0132][0133]
图8验证了本实施例中的带双键的酸响应性可去除聚乙二醇的成功制备。
[0134]
实施例3
[0135]
双重响应性高载药量聚前药的合成,所述方法具体包括以下步骤:
[0136]
将0.28g带双键的酸响应性可去除聚乙二醇、0.05三聚体结构的喜树碱前药单体、0.00434g链转移剂和0.00059gaibn(偶氮二异丁腈)溶解于1.5ml的dmso/三氯甲烷(体积比为4/1)混合溶剂中,液氮冻抽三次,在65℃下搅拌反应过夜。反应结束后,用二氯甲烷稀释后,从冷乙醚中沉淀,在室温下真空干燥,得到双重响应性高载药量聚前药,其结构式为:
[0137][0138]
所述的双重响应性高载药量聚前药的合成路线为:
[0139][0140]
如图9-10所示,图9为本实施例相应的核磁谱图,验证了本实施例-双重响应性高载药量聚前药的成功制备,图10的(a)、(b)分别为本发明实施例3中双重响应性高载药量聚前药的紫外谱图和荧光谱图,图10(a)中在370nm位置有喜树碱的特征吸收峰以及图10(b)中在430nm位置有喜树碱的特征发射峰,验证了本实施例中的双重响应性高载药量聚前药的成功制备。
[0141]
实施例4
[0142]
可变形双重响应性高载药量聚前药纳米载体的合成,包括以下步骤:
[0143]
取5mg双重响应性高载药量聚前药溶于1ml(四氢呋喃)中,缓慢滴入到剧烈搅拌的去离子水中(转速:1300-1500rpm,1h滴完),聚前药滴完后,保持高速(转速为1300-1500rpm)搅拌25-35min,之后低速(转速为300-500rpm)搅拌3h,转移到透析袋中用去离子水透析24h(约间隔4h换一次去离子水),得到球形结构的单分子胶束,即可变形双重响应性高载药量聚前药纳米载体。
[0144]
图11(a)为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体的动态光散射图,图11(b)为其在ph:5.0的酸处理前后的zeta电位图,验证了可变形双重响应性高载药量聚前药纳米载体的粒径为100-200nm,胶束外围带有微弱的负电荷,有利于延长纳米载体在体内的循环时间,增强细胞内吞和epr效应,且在ph:5.0的酸处理后负电荷大幅度
减少至几乎不带电荷,有利于纳米载体与细胞的结合,从而增强抗肿瘤功效。
[0145]
图12为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在ph(5.0)处理2小时、以及在dtt(15mm)处理6小时和24小时后的动态光散射图,验证了纳米载体在酸处理后尺寸变小了,经过dtt(15mm)处理后粒径逐渐减小,验证了纳米载体裂解了,进而释放出喜树碱药物,杀死肿瘤细胞。
[0146]
图13为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体的透射电子显微镜图,验证了可变形双重响应性高载药量聚前药纳米载体的粒径为100-200nm,有利于增强epr效应。
[0147]
图14为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在ph(5.0)处理2小时后的透射电子显微镜图,验证了纳米载体在酸处理后尺寸变小了。
[0148]
图15为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在dtt(15mm)处理24小时后的透射电子显微镜图,验证了纳米载体裂解了,进而释放出喜树碱药物,杀死肿瘤细胞。
[0149]
图16(a)、(b)分别为本发明实施例4中的可变形双重响应性高载药量聚前药纳米载体在dtt(15mm)作用下以及在dtt(15mm)和ph(5.0)的同时作用下,喜树碱原药分子释放曲线图,如图16所示,30h内两种情况下的喜树碱原药分子释放量都将近80%,说明纳米载体有很好的刺激响应性药物释放效果。
[0150]
以上内容仅仅是对本发明结构所作的举例和说明,所属本技术领域的技术人员对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,只要不偏离本发明的结构或者超越本权利要求书所定义的范围,均应属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1