一种高纯度波生坦的制备方法与流程
1.本发明属于药物化学技术领域,具体涉及一种高纯度波生坦的制备方法。
背景技术:
2.波生坦(bosentan)是由瑞典爱克泰隆(actelion pharmaceuticals ltd)公司和美国genentetch公司联合开发的一种特异性竞争性的双重内皮素受体阻滞剂。于2001年经fda批准用于原发性肺高压和硬皮病引起的肺高压。其结构式如下所示:
[0003][0004]
关于波生坦的合成方法,目前已报道的各种合成路线都是以4,6-二氯-5-(2-甲氧基-苯氧基)-2,2'-二嘧啶为基础,进行两步对氯基的取代反应最后得到波生坦,其中第一步取代反应基本都是用对叔丁基苯磺酰胺进行取代,主要是第二步羟乙基取代方法不同。
[0005]
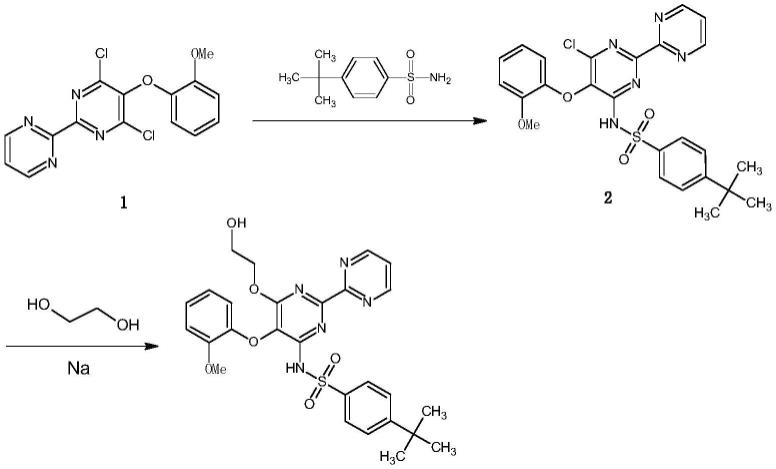
美国专利us005292740a最早报道了波生坦合成方法。该路线是以化合物1为起始原料,首先用对叔丁基苯磺酰胺进行取代,得到化合物2,然后加入乙二醇和金属钠反应得到波生坦。其反应式如下所示:
[0006][0007]
这条路线虽然比较短,很容易得到波生坦,但最大的缺点是使用了金属钠做碱,在工业生产中存在较大的安全隐患。并且该工艺的转化率较低,粗品中有大量副产物,难以精
制。
[0008]
美国专利us00613697a公开了波生坦的另一条制备路线,该路线是用其中一个羟基被叔丁基保护过的乙二醇与化合物2反应,生成化合物3,然后用甲酸替换保护基生成化合物4,最后用氢氧化钠处理得波生坦。其反应式如下所示:
[0009][0010]
这条路线最大的优势是理论上可以避免一分子乙二醇与两分子化合物2缩合而产生的副产物二聚体,但是由于去除保护基需要两步反应,延长了工艺路线,势必会提高成本和降低收率。
[0011]
美国专利us20100249162a1公开的波生坦的制备方法,是用氢氧化钠作催化剂和缚酸剂,使化合物2和乙二醇反应生成波生坦。合成路线如下所示:
[0012][0013]
这条路线反应条件温和,所用试剂友好,但是第一步反应收率低,另外该路线中涉及3个波生坦成品的主要杂质(杂质b、杂质d(反应物1)、杂质e(对叔丁基苯磺酰胺))含量较
9h,降温冷却,减压蒸馏;
[0023]
3)将蒸馏残留物加入l-酒石酸和水,保温反应1-2h,分离出固体,将所述固体加入二氯甲烷中,室温搅拌,加入吡啶和水,取有机层、浓缩至断流,加入醇和水的混合溶剂,析晶,得波生坦粗品;
[0024]
4)将波生坦粗品加入到醇和水的混合溶剂中,升温至回流,加入活性炭脱色0.5-1h,冷却析晶,得波生坦纯品,
[0025][0026]
优选的,上述本发明的制备方法,步骤1)中,式ⅰ化合物、4-叔丁基苯磺酰胺与缚酸剂的摩尔配比为1:(1.05-1.1):(1.1-1.2),式ⅰ化合物与甲苯的质量体积比(g/ml)为1:10-13,回流反应时间为4-10h,优选为为6-8h。所述室温优选为20℃,其中,所述的缚酸剂为碳酸钾或碳酸钠,优选为碳酸钾。
[0027]
优选的,上述本发明的制备方法,步骤2)中,式ⅱ化合物与四氢呋喃的质量体积比g/ml为1:6-8,式ⅱ化合物与乙二醇的质量体积g/ml为1:10-15,或者式ⅱ化合物与氢氧化钠的摩尔配比为1:8-10。
[0028]
优选的,上述本发明的制备方法,步骤3)中,所述保温反应,温度为60-65℃,式ⅱ化合物与l-酒石酸的摩尔比为1:4.0-5.0;步骤4)中,波生坦粗品和醇的质量体积比g/ml为1:8-15。
[0029]
优选的,上述本发明的制备方法,步骤3)中,后处理的分层试剂选自二氯甲烷、吡啶和水,三者的体积比为(7-8):1:(7-9)。
[0030]
优选的,上述本发明的制备方法,步骤3)和4)中,所述醇和水的混合溶剂,醇与水的体积比为6-8:1,其中,所述醇为甲醇或乙醇。
[0031]
在一具体实施方案中,本发明的一种高纯度波生坦的制备方法,包括如下步骤:
[0032]
1)将甲苯、4-叔丁基苯磺酰胺和缚酸剂碳酸钾或碳酸钠混合搅拌2-4h,后加入式ⅰ化合物,升温至回流反应4-10h,得化合物ⅱ;
[0033]
2)将乙二醇、氢氧化钠和四氢呋喃混合、搅拌、溶清,加入式ⅱ化合物,升温回流8-9h,降温冷却,减压蒸馏;
[0034]
3)将蒸馏残留物加入l-酒石酸和水,保温反应1-2h,分离固体,将所得固体加入二氯甲烷中,室温搅拌,加入吡啶和水,分出有机层、将有机层浓缩至断流,再加入醇和水的混合溶剂,析晶,得波生坦粗品;
[0035]
4)将波生坦粗品加入到醇和水的混合溶剂中,升温至回流,加入活性炭脱色0.5-1h,冷却析晶,得波生坦纯品。
[0036]
在上述具体实施方案中,优选的,本发明所述的波生坦制备方法,步骤1)中,所述缚酸剂为碳酸钾,4-叔丁基苯磺酰胺与缚酸剂碳酸钾混合搅拌时间为2h,回流反应时间为6-8h。
[0037]
在上述具体实施方案中,优选的,本发明所述的波生坦制备方法,步骤1)中,式ⅰ化合物与甲苯的质量体积比(g/ml)为1:10-13,式ⅰ化合物与4-叔丁基苯磺酰胺、碳酸钾的摩尔配比为1:(1.05-1.1):(1.1-1.2)。
[0038]
在上述具体实施方案中,进一步包括,将步骤1)回流反应所得式ⅱ化合物在用醇溶剂结晶,优选地,所述醇溶剂为甲醇。
[0039]
在上述具体实施方案中,优选的,本发明的波生坦制备方法,步骤2)中,式ⅱ化合物与四氢呋喃的质量体积比(g/ml)为1:6-8,与乙二醇的质量体积比(g/ml)为1:10-15,与氢氧化钠的摩尔配比为1:8-10。
[0040]
在上述具体实施方案中,优选的,本发明的波生坦制备方法,步骤3)中,所述保温反应,其反应温度为60-65℃,式ⅱ化合物与l-酒石酸的摩尔比为1:4.0-5.0,所述醇和水的混合溶剂,醇和水的体积比为6-8:1。
[0041]
在上述具体实施方案中,优选的,本发明所述的波生坦制备方法,步骤3)中,后处理的分层试剂选自二氯甲烷、吡啶和水,三者的体积比为(7-8):1:(7-9)。二氯甲烷、吡啶、水三种溶剂作为分层提取的溶剂,其重量比是影响产品纯度能达到98.5%以上的重要因素之一。
[0042]
在上述具体实施方案中,优选的,步骤4)中波生坦粗品和醇的质量体积比(g/ml)为1:8-15,所述醇和水的混合溶剂,醇和水的体积比为6-8:1,析晶温度为20-25℃,波生坦干燥温度为50-65℃,干燥时间为4-6h。
[0043]
在上述具体实施方案中,优选的,步骤3)和步骤4)中所述得醇选自甲醇或乙醇,优选的,为甲醇。
[0044]
本发明所提供的波生坦的制备工艺,操作简便可控,有效提高了反应转化率,目标产物波生坦的纯度高,降低杂质含量,尤其是成品中有关物质b、c、d、e的含量得到极好控制,是一条适合于工业化生产的制备方法。
具体实施方式
[0045]
以下实施例仅是代表性的,用于进一步阐明和理解本发明的实质,但不以任何方式限制本发明的范围。
[0046]
实施例1波生坦的制备
[0047]
第一步:将300ml甲苯、17.5g 4-叔丁基苯磺酰胺和12.5g研磨后的碳酸钾加入反应瓶,保温20℃,搅拌2h,然后加入27g化合物ⅰ(即式i化合物),升温至回流(130℃左右)反应6h。将反应液冷却至50℃以下,抽滤,所得滤液减压蒸馏。在蒸馏残留物中加入400ml二氯
甲烷和400ml饮用水,搅拌冷却至5~10℃,用稀盐酸20g(浓盐酸10g,饮用水10g)调ph至3.0~4.0。分出水层,二氯甲烷提取两次,合并有机层,向有机层中加入无水硫酸钠50g,干燥0.5小时,过滤,滤液减压蒸馏,得油状物,加入160ml甲醇于20~30℃搅拌析晶1h,抽滤,将所得湿品于40~50℃干燥4小时,得化合物ⅱ(即式ii化合物)35.2g,收率86.6%。
[0048]
第二步:将360ml乙二醇,22g naoh和220ml四氢呋喃加入反应瓶,搅拌升温至45~50℃。待naoh溶清后,加入第一步所得的化合物ⅱ35.0g,升温至60~65℃反应8小时,浓缩。将所得残留物冷却至10℃。
[0049]
第三步:残留物加入45g l-酒石酸和120ml水,保温搅拌1.5h。抽滤,滤饼用120ml水洗1次,所得固体于55~60℃,减压干燥4h,得类白色固体30.5g,加入到105ml二氯甲烷中,室温搅拌溶解,再加入吡啶15ml,水120ml,搅拌1小时,分出水层,有机层减压浓缩,所得浓缩液中加入160ml甲醇、25ml水的混合液,搅拌析晶1h,抽滤,将所得固体于40~50℃干燥6h,得类白色固体33.3g,收率88.0%。
[0050]
第四步:将上步所得的波生坦粗品30g加入到280g甲醇和40g纯化水中,升温至60℃,溶解完全,加入活性炭3g,搅拌0.5h,过滤除去活性炭,将滤液冷却至20~25℃,搅拌析晶2h,过滤,50-55℃干燥2h,得波生坦纯品29.0g,收率96%。hplc纯度99.50%。1h-nmr(400mhz,cdcl3)δ:9.60(d,2h),8.30(t,2h),8.23(t,3h),7.31(d,2h),7.01-7.10(m,1h),6.90(t,1h),6.78-6.80(m,2h),4.85(t,2h),3.76-3.79(m,5h),1.20(m,9h)。
[0051]
实施例2波生坦的制备
[0052]
第一步:将300ml甲苯、17.5g 4-叔丁基苯磺酰胺和12.5g研磨后的碳酸钠加入反应瓶,保温20℃搅拌2h,然后加入27g化合物ⅰ,升温至回流(130℃左右)反应6h。将反应液冷却至30~40℃,抽滤,所得滤液减压蒸馏。在残留物中加入400ml二氯甲烷和400ml饮用水,搅拌冷却至5~10℃,用稀盐酸20g(浓盐酸10g,饮用水10g)调ph至3.0~4.0。分出水层,二氯甲烷提取两次,合并有机层,向有机层中加入无水硫酸钠50g,干燥0.5小时,过滤,滤液减压蒸馏,得油状物,加入160ml甲醇于20~30℃搅拌析晶1h,抽滤,将所得湿品于40~50℃干燥4小时,得化合物ⅱ34.3g,收率84.3%。
[0053]
第二步:将360ml乙二醇,22g naoh和210ml四氢呋喃加入反应瓶,搅拌升温至48~50℃。待naoh溶清后,加入第一步所得的化合物ⅱ34.0g,升温至65℃反应8小时,浓缩。将所得残留物冷却至10℃。
[0054]
第三步:残留物加入47g l-酒石酸和120ml水,保温搅拌1.5h。抽滤,滤饼用120ml水洗1次,所得固体于55~60℃,干燥4h得,类白色固体30.5g。加入到120ml二氯甲烷中,室温搅拌溶解,再加入吡啶15ml,水135ml,搅拌1小时,分出水层,有机层减压浓缩,所得浓缩液中加入160ml甲醇、25ml水的混合液,搅拌析晶1h,抽滤,将所得固体于40~50℃,真空度0.07~0.09mpa下减压干燥6h,得类白色固体32.94g,收率87%。
[0055]
第四步:将第三步所得的29g波生坦粗品加入到280g乙醇和40g纯化水中,升温至60℃,溶解完全,加入活性炭3g,搅拌0.5h,过滤除去活性炭,将滤液冷却至20~25℃,搅拌析晶2h,过滤,50-55℃干燥2h,得波生坦纯品27.5g,收率94.8%。
[0056]
对比例1
[0057]
参照专利cn102421770a中实施例4的方法制备式ii化合物。
[0058]
对比例2
[0059]
参照实施例1的方法,将300ml甲苯、17.5g 4-叔丁基苯磺酰胺和12.5g研磨后的碳酸钾加入反应瓶,搅拌,然后加入27g化合物ⅰ(即式i化合物),升温至回流(130℃左右)反应6h。将反应液冷却至50℃以下,抽滤,所得滤液减压蒸馏。在蒸馏残留物中加入400ml二氯甲烷和400ml饮用水,搅拌冷却至5~10℃,用稀盐酸20g(浓盐酸10g,饮用水10g)调ph至3.0~4.0。分出水层,二氯甲烷提取两次,合并有机层,向有机层中加入无水硫酸钠50g,干燥0.5小时,过滤,滤液减压蒸馏,得油状物,加入160ml甲醇于20~30℃搅拌析晶1h,抽滤,将所得湿品于40~50℃干燥4小时,得化合物ⅱ(即式ii化合物)32.6g,收率80.2%。
[0060]
对比例3
[0061]
在实施例的基础上,将步骤三中的二氯甲烷、吡啶和水的量变分别为150ml、15ml和150ml或者75ml、15ml和90ml。其它都不变,制得化合物ⅱ,分别为28.1g和27.6g,纯度分别为97.5%和97.7%。
[0062]
实施例3杂质和纯度测定
[0063]
测定实施例1和实施例2获得的波生坦,与美国us 20100249162a1公开的方法制得的波生坦测定的纯度及有关物质,并进行对比研究,如下表1所示:
[0064]
表1实施例1、2及美国专利制得的波生坦的纯度和有关物质
[0065][0066]
杂质a结构式为:
[0067][0068]
实施例4式ii化合物的纯度和有关物质含量测定
[0069]
检测实施例1和2的步骤1)制得的式ii化合物和对比例1和2制得的式ii化合物的纯度和有关物质,其结果见表2。
[0070]
表2中间体式‖化合物的纯度和有关物质的含量
[0071][0072]
表2的结果表明,实施例1和2的中间体式ii化合物与比例1和2的中间体式ii化合物无论是在纯度还是在有关无物质方面都表现出优势,特别是杂质d和e的含量优势特别明显。本研究发现,碳酸钾一方面是缚酸剂,在反应进行过程中生成的hcl自然会被它中和,另外作为催化剂的作用,需要使用碱性物质让对叔丁基苯磺酰胺的氨基氢充分活化后才能实现,因此将对氨基苯磺酰胺先和碳酸钾先反应一段时间,使氨基上的酸性氢在碱性作用下充分活化,提供质子的能力加强,更易于上化合物i上的氯生成hcl,有利于提高转化和降低杂质。
[0073]
以上实施例是典型的,任何在本发明的精神实质的基础上进行简单的变化和修饰变通都属于本发明的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1