一类泛-KRAS抑制剂及其制备和应用的制作方法
一类泛-kras抑制剂及其制备和应用
技术领域
1.本技术涉及一类四氢异喹啉衍生物,其制备方法,含有这些化合物的药物组合物或其盐以及作为泛-kras抑制剂在治疗不同肿瘤中的医学用途。
背景技术:
2.ras是首个被发现的人类肿瘤基因(oncogene),是肿瘤中最常见的突变基因之一,在约30%的肿瘤中均携带有ras突变,如果结合ras的调控因子和信号通路的上下游突变,则几乎覆盖所有肿瘤。kras基因(kirsten rat sarcoma viral oncogene homolog)是ras基因家族中的重要成员。kras基因编码的蛋白是gdp/gtp结合蛋白,是一种小gtpase酶,它属于超蛋白家族。kras蛋白有188个氨基酸,其分子量为21.6kd,其定位于细胞膜内侧,通过法尼酰基(farnesyl)的修饰基因连接到细胞膜上。kras与gtp结合呈激活状态(kras-gtp),与gdp结合呈关闭状态(或非活状态)(kras-gdp),随后,gtp酶激活蛋白(gap)可以将结合在kras-gtp上的gtp水解为gdp,促使kras-gdp关闭状态的形成,从而使kras处在失活态。kras蛋白是处在kras-gtp激活状态和kras-gdp非活状态(关闭状态)之间的“开关”,在激活状态可激活下游信号通路其中包括mapk信号通路,pi3k信号通路和ral-gds信号通路。ras蛋白开关控制着其下游信号通路,从而促进细胞生存,增殖和细胞因子释放,在细胞增殖,分化和凋亡等生命过程中发挥着重要作用。kras也可被生长因子(如egfr)短暂激活,活化后的kras可激活下游如控制细胞生成的pi3k-akt-mtor信号通路,以及控制细胞增殖的ras-raf-mek-erk信号通路,而突变的kras即使没有egfr等激酶激活的情况下却会发生持续活化,导致细胞持续增值,最终发生癌变。
3.kras突变在多种肿瘤中高表达,被发现到最常见的包括肺癌,肠癌,胰腺癌、结肠癌、小肠癌、胆管癌等。结构学研究表明,kras的基因突变大多干扰了kras水解gtp的能力,最终使kras持续激活,使之无法有效调控细胞信号转导,从而促进肿瘤的发生、发展以及转移。
4.对于kras突变,12位氨基酸(g12)的突变约占80%,而g12c突变大约占g12全部突变的14%。近几年来,研究人员相继开发了一系列kras g12c突变共价抑制剂,但开发kras g12d突变抑制剂遇到了极大的挑战。
5.目前还没有开发出共价结合在天冬氨酸的方法。直接抑制kras g12d突变体难点不仅在于kras编码的蛋白表面光滑,缺少结合位点,且kras与gtp/gdp的结合力非常强,胞内gtp/gdp的浓度也很高,导致无法开发对gtp竞争性抑制剂。不仅kras膜定位受法尼基转移酶等调节,而且靶向kras下游信号分子(效应蛋白),抑制生长所需的野生型信号通路的治疗窗口狭小,更由于补偿机制使无法完全而有效地抑制kras突变体下游信号,从而使开发效应蛋白的激酶抑制剂对kras突变的疗效受到极大限制。
6.综上所述,对于开发具有口服安全有效性的泛-kras抑制剂仍然有很大的未能满足的临床需求。
技术实现要素:
7.本发明的目的是提供一种具有口服安全有效性的泛-kras抑制剂,特别是用于治疗肠癌、肺癌、胰腺癌、胆管癌、胃癌等肿瘤的治疗的抑制剂。
8.本发明的第一方面,提供了一种如下式(i)所示的化合物,或其药学上可接受的盐:
[0009][0010]
其中,
[0011]
m为1、2、3、4或5;
[0012]
n为1、2或3;
[0013]
r1选自下组:氢、卤素、oh、sh、nh2、取代或未取代的c
1-6
烷基;
[0014]
r2选自下组:取代或未取代的c
1-6
烷基、取代或未取代的c
1-6
烷氧基;
[0015]
r3选自下组:氢、卤素、取代或未取代的c
1-6
烷基;
[0016]
x选自下组:o、n、nh、s、ch、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为h或c
1-4
烷基;
[0017]
y选自n、nh、ch、ch2、ch2ch2、cf2、cr8、or9、sr
10
;
[0018]
z选自o、n、nh、s、ch、ch2、cf2、cfh、nr
12
;
[0019]
r8为c
1-5
烷基;
[0020]
r9为c
1-5
烷基;
[0021]r10
为c
1-5
烷基;
[0022]
虚线为化学键或无。
[0023]
在另一优选例中,所述的式(i)化合物具有如下式(ii)所示的结构:
[0024][0025]
其中,
[0026]
m为1、2、3、4或5;
[0027]
n为1、2或3;
[0028]
r1选自下组:氢、卤素、oh、sh、nh2、取代或未取代的c
1-6
烷基;
[0029]
r2选自下组:取代或未取代的c
1-6
烷基、取代或未取代的c
1-6
烷氧基;
[0030]
r3选自下组:氢、卤素、取代或未取代的c
1-6
烷基;
[0031]
a选自下组:o、n、nh、s、ch、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为h或c
1-4
烷基;
[0032]
b选自o、n、nh、s、ch、ch2、cf2、cfh、nr
12
。
[0033]
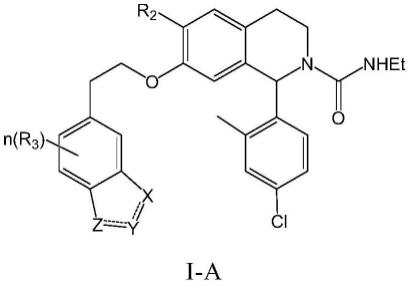
在另一优选例中,所述的式(i)化合物具有如下式(i-a)的结构:
[0034][0035]
其中,
[0036]
n为1、2或3;
[0037]
r2选自下组:卤素,取代或未取代的c
1-6
烷基、取代或未取代的c
1-6
烷氧基;
[0038]
r3选自下组:氢、卤素、取代或未取代的c
1-6
烷基;
[0039]
x选自下组:o、n、nh、s、ch、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为h或c
1-4
烷基;
[0040]
y选自n、nh,ch,ch2、ch2ch2、cf2、cr8、or9、sr
10
;
[0041]
z选自o、n、nh、s、ch、ch2、cf2、cfh、nr
12
;
[0042]
r8为c
1-5
烷基;
[0043]
r9为c
1-5
烷基;
[0044]r10
为c
1-5
烷基。
[0045]
在另一优选例中,所述的式(i)化合物具有如下式(ii-a)的结构:
[0046][0047]
其中,
[0048]
n为1、2或3;
[0049]
r2选自下组:取代或未取代的c
1-4
烷基,取代或未取代的c
1-4
烷氧基;
[0050]
r3选自下组:氢、卤素、取代或未取代的c
1-4
烷基;
[0051]
a选自下组:o、n、nh、s、ch、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为h或c
1-4
烷基;
[0052]
b选自o、n、nh、s、ch、ch2、cf2、cfh、nr
12
。
[0053]
|[31]4、如权利要求1所述的化合物,或其药学上可接受的盐,其特征在于,所述的r2为c
1-4
烷氧基。
[0054]
在另一优选例中,所述的x选自下组:o、nh、s、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为c
1-4
烷基;
[0055]
y选自ch、n;
[0056]
z选自n或ch。
[0057]
在另一优选例中,所述的x选自下组:n或ch;
[0058]
y选自ch、n;
[0059]
z选自o、nh、s、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为c
1-4
烷基。
[0060]
在另一优选例中,所述的x选自下组:o、nh、s、ch2、cf2、cfh、nr
12
;
[0061]
y选自下组:ch2、cf2、cfh;
[0062]
z选自下组:o、nh、s、ch2、cf2、cfh、nr
12
;其中,所述的r
12
为c
1-4
烷基。
[0063]
在另一优选例中,所述的a为o,且所述的b为nh;且r3为h。
[0064]
本发明的第二方面,提供了如本发明第一方面所述的化合物的用途,其用于制备治疗与kras突变体活性或表达量相关的疾病的药物的用途。
[0065]
在另一优选例中,所述与kras突变体活性或表达量相关的疾病为肿瘤,较佳地为选自下组的肿瘤:肉瘤、粘液瘤、横纹肌瘤、纤维瘤、脂肪瘤、畸胎瘤、支气管癌、肺癌、支气管腺瘤、淋巴瘤、软骨瘤错构瘤、间皮瘤、食道癌、胃癌、胰腺癌、小肠癌、大肠癌、泌尿生殖道肿瘤、肾癌、膀胱癌、尿道癌、前列腺、睾丸癌、肝癌、胆管癌、肝母细胞瘤、血管肉瘤、肝细胞腺瘤、血管瘤、胆囊癌、壶腹癌、胆管癌、骨癌、脑癌、子宫癌、阴道癌、血液瘤、皮肤癌、乳腺癌。
[0066]
本发明的第三方面,提供了一种药物组合物,所述的药物组合物包括:(i)治疗有效量的如本发明第一方面所述的式i化合物,或其药学上可接受的盐;和(ii)药学上可接受的载体。
[0067]
在另一优选例中,所述的有效量是指治疗有效量或抑制有效量,较佳地为0.01~99.99%。
[0068]
在另一优选例中,所述的药物组合物用于治疗与kras突变体活性或表达量相关的疾病。
[0069]
在另一优选例中,所述kras突变体为kras g12d突变体、kras g12v突变体、kras g12s突变体或kras g13d突变体。
[0070]
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
具体实施方式
[0071]
本发明人经过长期而深入的研究,制备了一类具有式i所示结构的化合物,并发现其具有抑制kras-effector蛋白-蛋白相互作用的活性,是一种泛-kras抑制剂。基于上述发现,发明人完成了本发明。
[0072]
术语
[0073]
如本文所用,术语“c
1-c6烷基”指具有1~6个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基,或类似基团,“c
1-c3烷基”等表述具有类似的定义。
[0074]
术语“c
1-c6烷氧基”指具有1~6个碳原子的直链或支链烷氧基,例如甲氧基、乙氧基、丙氧基、异丙氧基,或类似基团,“c
1-c3烷氧基”等表述具有类似的定义。
[0075]
本发明中,术语“含有”、“包含”或“包括”表示各种成分可一起应用于本发明的混合物或组合物中。因此,术语“主要由...组成”和“由...组成”包含在术语“含有”中。
[0076]
本发明中,术语“药学上可接受的”成分是指适用于人和/或动物而无过度不良副反应(如毒性、刺激和变态反应),即有合理的效益/风险比的物质。
[0077]
本发明中,术语“有效量”指治疗剂治疗、缓解或预防目标疾病或状况的量,或是表现出可检测的治疗或预防效果的量。对于某一对象的精确有效量取决于该对象的体型和健康状况、病症的性质和程度、以及选择给予的治疗剂和/或治疗剂的组合。因此,预先指定准确的有效量是没用的。然而,对于某给定的状况而言,可以用常规实验来确定该有效量,临床医师是能够判断出来的。
[0078]
在本文中,除特别说明之处,术语“取代”指基团上的一个或多个氢原子被选自下组的取代基取代:卤素、未取代或卤代的c
1-c6烷基、未取代或卤代的c
2-c6酰基、未取代或卤代的c
1-c6烷基-羟基。
[0079]
除非特别说明,本发明中,所有出现的化合物均意在包括所有可能的光学异构体,如单一手性的化合物,或各种不同手性化合物的混合物(即外消旋体)。本发明的所有化合物之中,各手性碳原子可以任选地为r构型或s构型,或r构型和s构型的混合物。
[0080]
术语“环烷基”包括具有3至12个碳,例如3至8个碳,并且作为进一步实例3至6个碳的饱和和部分不饱和的环烃基,其中所述环烷基另外任选地被一个或多个取代。环烷基的实例包括但不限于环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环庚基和环辛基。术语“环烷基”还包括桥连环烷基,例如双环[1.1.1]戊基。
[0081]
如本文所用,术语“芳基”基团是包含一到三个芳环的c6-c14芳族部分。作为一个实施例,芳基是c6-c10芳基。芳基的实例包括但不限于苯基、萘基、蒽基、芴基和二氢苯并呋喃基。“芳基”还指二环或三环环系统,其中所述芳环系统的一个或两个环分别可以是饱和或部分饱和的,并且其中如果所述环系统包括两个饱和环,则所述饱和环可以是稠合的或螺环,但其与化合物的其他部分的连接位置在芳基部分上。
[0082]“杂环基”或“杂环”基团是具有3至12个原子,例如4至8个原子的环结构,其中一个或多个原子选自由n、o和s组成的组,其中环n原子可以被氧化成no,并且环s原子可以被氧化成so或so2,其余的环原子是碳。杂环基可以是单环、双环、螺环或桥环系统。
[0083]
术语“杂芳基”是指具有5至14个环原子,优选5、6、9或10个环原子的基团;并且除碳原子外,每个环具有一至三个选自n、o和s的杂原子,“杂芳基”还指除碳原子外,每个环具有一到三个选自n、o和s的杂原子的双环系统,其中一个环系统可以是饱和的或部分饱和的。
[0084]
术语“卤素”指f、cl、br和i。
[0085]
如本文所用,术语“本发明化合物”指式i所示的化合物。该术语还包括及式i化合物的各种晶型形式、药学上可接受的盐、水合物或溶剂合物。
[0086]
如本文所用,术语“药学上可接受的盐”指本发明化合物与酸或碱所形成的适合用
作药物的盐。药学上可接受的盐包括无机盐和有机盐。一类优选的盐是本发明化合物与酸形成的盐。适合形成盐的酸包括但并不限于:盐酸、氢溴酸、氢氟酸、硫酸、硝酸、磷酸等无机酸,甲酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、苦味酸、甲磺酸、苯甲磺酸,苯磺酸等有机酸;以及天冬氨酸、谷氨酸等酸性氨基酸。
[0087]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
[0088]
制备例1
[0089][0090]
实验步骤:
[0091]
步骤a:
[0092]
室温条件下,将原料1-1(30.0克),1-2(47.0克),三氟乙酸126ml混合后,反应液在80℃搅拌16小时。lcms显示检测到反应完毕,反应液在真空中蒸发得到粗产品。添加200毫升冰水液体,然后加入naoh,将液体中和到ph值为8-9,并用dcm(500毫升*2)萃取。合并后的有机相经饱和食盐水(300毫升)洗涤。将有机层在无水na2so4结晶干燥,过滤后在真空中蒸发得到粗产品,通过硅胶过柱纯化(meoh:dcm:=0.5-2.5%)得到白色固体1-3(24.1克)。
[0093]
lcms:(esi)m/z 304.1(m+h)
+
.
[0094]
步骤b:
[0095]
室温条件下,将1-3(20.0克),tea(18.40毫升)溶于thf(200毫升),滴加乙基异氰酸酯(7.83毫升)。反应液在25℃搅拌1小时。过滤后在真空中蒸发得到粗产品。粗品用硅胶柱层析纯化(乙酸乙酯:石油醚=5%-10%),得到白色固体制备例1(7.0克)。
[0096]
ms(esi)m/z:375.3[m+h]
+
。
[0097]
实施例1
[0098][0099]
实验步骤:
[0100]
步骤a:
[0101]
0度温度条件下,将2-氨基-5-溴苯酚1-1(1.0克)和碳酸氢钠(1.34克)溶于1,2-二甲氧基乙烷(25毫升)和水(2毫升中)后搅拌下滴加2-氯乙酰氯(902毫克)。添加后的反应混合物在同样温度下搅拌30分钟后,在80度加热搅拌16小时。冷却到室温后,将反应混合物倒入冰水(100毫升),所得固体经过滤收集,冻干后得到浅棕色固体1-2(1.05克)。
[0102]1h nmr(400mhz,dmso-d6)δ10.81(s,1h),7.17-7.12(m,2h),6.83(d,j=8.4hz,1h),4.60(s,2h).
[0103]
步骤b
[0104]
在0度下将硼烷的四氢呋喃溶液(17.5毫升,浓度为1.0摩尔)滴加到原料1-2(2.0
克)的thf(10毫升)溶液中。反应混合物在70度搅拌3小时。反应物冷却后在0度搅拌下滴加甲醇(2毫升)。反应混合物减压浓缩后粗品用硅胶柱层析纯化(乙酸乙酯:石油醚=3/1),得到浅棕色固体1-3(1。81克)。
[0105]
ms:m/z 214.1[m+h]
+
.
[0106]
步骤c
[0107]
将原料1-3(1.8克),三乙基胺(1.7可)和二碳酸二叔丁酯(2.75克)溶于二氯甲烷(50毫升),反应液在25℃搅拌16小时。反应完全后减压浓缩后粗品用硅胶柱层析纯化(乙酸乙酯:石油醚=5/1),得到白色固体1-4(2.40克)。
[0108]
lcms:m/z 213.9[m+h-100]
+
.
[0109]
步骤d
[0110]
氩气保护下将1-4(1.78克,5.69毫摩尔),(e)-1-乙氧乙烯基-2-硼酸频那醇酯(1.69克,8.53毫摩尔),1,1'-双(二苯基膦)二茂铁二氯化钯((416毫克,0.569毫摩尔),碳酸钾(2.35克,17.06毫摩尔),1,4-二氧六环(40毫升)和水(10毫升)的混合物加热至90℃搅拌2小时。将反应液冷却至室温后经过滤后加水(10毫升)并用乙酸乙酯(3x 20毫升)萃取。合并后的有机相经干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯20/1-21),得到浅黄色固体1-5(1.70克)。
[0111]
lcms:m/z 250.0[m+h-56]
+
.
[0112]
步骤e和f:
[0113]
将中间体1-5(500毫克)溶于二氯甲烷(10毫升),加入甲酸(1毫升)。反应液在40℃搅拌16小时。薄层硅胶柱层析检测反应完全后,混合物用饱和碳酸氢钠水溶液(50毫升)中和,用乙酸乙酯(50毫升)萃取三次。结合有机层,加入饱和食盐水(30毫升)洗涤,有机层用无水硫酸钠干燥。处理后反应液经减压浓缩得到粗产品中间体,不经分离直接用于下一步反应。
[0114]
在0度下将硼氢化钠(125毫克,3.28毫摩尔)加入到上述粗产品中间体(1.0克)的甲醇(20毫升)溶液中。反应混合物室温搅拌3小时。反应结束后加水(50毫升),用乙酸乙酯(50毫升)萃取三次。结合有机层,加入饱和食盐水(30毫升)洗涤,有机层用无水硫酸钠干燥。处理后反应液经减压浓缩得到粗产品中间体,反应混合物减压浓缩后粗品用硅胶柱层析纯化(乙酸乙酯:石油醚=3/1),得到黄色油状物1-6(304毫克)。
[0115]1h nmr(400mhz,cdcl3):δ7.71(s,1h),6.75-6.73(m,2h),4.23(d,j=4.6hz,2h),3.84(d,j=4.6hz,4h),2.78(d,j=6.6hz,2h),1.54(s,9h).
[0116]
步骤g
[0117]
室温下将1-6(150毫克,0.53毫摩尔),三苯基膦(182mg,0.7毫摩尔),制备例1(200mg,0.53毫摩尔)的甲苯(10ml)溶液氩气抽换气三次,氩气保护下加热至120℃搅拌10分钟。随后逐滴加入偶氮二甲酸二异丙酯(0.14ml,0.70毫摩尔)。反应液随后120℃并且氩气保护下搅拌2小时。反应液冷却至室温后加水(50ml),所得混合物用乙酸乙酯(3x50毫升)萃取。合并后的有机相经饱和食盐水(30毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯1/1)得到白色固体1-7(196毫克)。
[0118]
lcms:m/z 636.3[m+h]
+
.
[0119]
步骤h
[0120]
将中间体1-7(196毫克,0.31毫摩尔)溶于二氯甲烷(10毫升),加入三氟乙酸(1毫升)。反应液在25℃搅拌16小时。反应完全后,混合物加入碳酸氢钠水溶液(20毫升)中和,用乙酸乙酯(50毫升)萃取三次。结合有机层,加入饱和食盐水(30毫升)洗涤,有机层用无水硫酸钠干燥。处理后反应液经减压浓缩得到粗产品,用制备色谱柱(waters2767/2545/2489/qda,inertsil ods-3 10um 20*250nm,流动相a:0.1%甲酸水溶液,流动相b:乙腈,流速:20毫升/m分钟:柱温:室温)分离得到白色固体实施例1(63.56毫克)。
[0121]
ms(esi)m/z:536.3[m+h]
+
;
[0122]1h nmr(400mhz,dmso-d6)δ7.25(d,j=2.0hz,1h),7.08(dd,j=8.4hz,1h),6.76(s,1h),6.67(t,j=5.4hz,1h),6.54
–
6.49(m,5h),6.44(d,j=7.6hz,1h),6.54(s,1h),4.07(t,j=4.2hz,2h),3.96
–
3.90(m,1h),3.83
–
3.76(m,1h),3.75(s,3h),3.73
–
3.69(m,1h),3.22(t,j=4.0hz,2h),3.10
–
3.08(m,2h),2.97
–
2.80(m,2h),2.74(t,j=7.4hz,2h),2.57
–
2.54(m,1h),2.40(s,3h),1.01(t,j=7.2hz,3h).
[0123]
实施例2
[0124][0125]
实施例2的制备按照类似实施例6的方法得到。用2-(苯并[d]恶唑-5-基)乙烷-1-醇(30mg)和制备例1(69毫克)得到白色固体实施例3(33.12毫克)。
[0126]
ms(esi):m/z 520.2[m+h]
+
.
[0127]1h nmr(400mhz,dmso-d6)δ8.69(s,1h),7.72(s,1h),7.65(d,j=8.0hz,1h),7.35
–
7.33(m,1h),7.23(d,j=2.0hz,1h),7.10
–
7.07(m,1h),6.77(s,1h),6.69
–
6.66(m,1h),6.54
–
6.48(m,3h),4.11
–
4.06(m,1h),4.01
–
3.97(m,1h),3.76(s,3h),3.74
–
3.69(m,1h),3.11
–
3.04(m,4h),2.95
–
2.78(m,2h),2.57
–
2.56(m,1h),2.39(s,3h),1.00(t,j=7.2hz,3h).
[0128]
实施例3
[0129][0130]
步骤a:
[0131]
将2-(3-氨基-4-羟基苯基)乙酸甲酯3-1(2.00克,11.0毫摩尔)溶于乙酸乙酯(4毫升)和水(4毫升)中,0度下加入碳酸氢钠(1.00克,12.1毫摩尔)和氯乙酰氯(1.42毫升,11.0毫摩尔),室温下搅拌反应3小时。tlc监测反应结束后,反应液用乙酸乙酯(20毫升)和盐水(20毫升)稀释。分离有机层,无水硫酸钠干燥,过滤并浓缩。所得残渣溶于n,n-二甲基甲酰胺(50毫升),加入碳酸钾(3.36克,22.0毫升)。反应加热至80度,继续搅拌反应3小时。tlc监测反应结束后,反应冷却至室温,用水(100毫升)稀释,乙酸乙酯(100毫升
×
3)萃取,合并有机相,用盐水(50毫升)洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩得到2-(3-氧代-3,4-二氢-2h-苯并[b][1,4]恶嗪-6-基)乙酸甲酯3-2(1,7克,黄色油状产品)。
[0132]
ms(esi):m/z 222.1[m+h]
+
[0133]
步骤b:
[0134]
在0度下,向2-(3-氧代-3,4-二氢-2h-苯并[b-[1,4]恶嗪-6-基)乙酸酯3-2(1.70克,7.70毫摩尔)的四氢呋喃(20毫升)溶液中分批加入氢化铝锂(0.73克,19.3毫摩尔),室温下搅拌反应过夜。lc-ms监测反应结束后,将所得混合物用水(0.8毫升)淬灭,然后0度下加入氢氧化钠水溶液(0.8毫升,3.0摩尔浓度)和水(2.4毫升),所得混合物用硅藻土过滤,滤液浓缩,所得残余物用硅胶柱层析法(二氯甲烷/甲醇=100/1至20/1)纯化得到2-(3,4-二氢-2h-苯并[b][1,4]恶嗪-6-基)乙-1-醇3-3(500mg,黄色油状物)。
[0135]
ms(esi):m/z 180.1[m+h]
+
[0136]
步骤c:
[0137]
向2-(3,4-二氢-2h-苯并[b][1,4]恶嗪-6-基)乙-1-醇3-3(500毫克,2.79毫摩尔)的二氯甲烷(20毫升)溶液中加入三乙胺(1.10毫升,8.40毫摩尔),二碳酸二叔丁酯(730毫克,3.38毫摩尔)和4-二甲氨基吡啶(100毫克,0.820毫摩尔),室温搅拌反应过夜。lc-ms监测反应结束后,反应液真空浓缩,所得残余物用硅胶柱层析法(石油醚/乙酸乙酯=4/1-1/1)纯化得到6-(2-羟乙基)-2,3-二氢-4h-苯并[b][1,4]恶嗪-4-羧酸叔丁酯3-4(80mg,白色固体),产率:10.2%。
[0138]
ms(esi):m/z 180.1[m-boc+h]
+
[0139]
步骤d:
[0140]
向6-(2-羟乙基)-2,3-二氢-4h-苯并[b][1,4]恶嗪-4-羧酸叔丁酯3-4(80毫克,0.286毫摩尔)的甲苯(5毫升)溶液中依次加入制备例1(107毫升,0.286毫摩尔)和三苯基膦(90毫克,0.343毫摩尔)。氩气保护下,反应混合物加热至105度,搅拌30分钟,加入偶氮二甲酸二异丙酯(70毫克,0.343毫摩尔),继续搅拌反应3小时。lc-ms监测反应结束后,反应冷却至室温,盐水(20毫升)稀释,乙酸乙酯(20毫升
×
3)萃取,无水硫酸钠干燥,过滤并减压浓缩,所得残余物用硅胶柱(石油醚/乙酸乙酯=1/1)纯化得到6-(2-((1-(4-氯-2-甲基苯基)-2-(乙基氨基甲酰基)-6-甲氧基-1,2-四氢异喹啉-7-基)氧基)乙基)-2,3-二氢-4h-苯并[b][1,4]恶嗪-4-羧酸叔丁酯3-5(150mg,黄色油状物)。
[0141]
ms(esi):m/z 658.3[m+na]
+
[0142]
步骤e:
[0143]
将3-5(150毫克,0.236毫摩尔)和盐酸/二氧六环(5毫升,4.0摩尔浓度)的混合物在室温下搅拌2小时。lc-ms监测反应结束后,反应液真空浓缩,所得残余物通过高效液相制备(nh 4hco 3)纯化得到实施例3(13.83mg,白色固体)。
[0144]
ms(esi):m/z 536.3[m+h]
+
[0145]1h nmr(400mhz,cdcl3)δ7.17(d,j=1.8hz,1h),6.97(dd,j=8.3,1.9hz,1h),6.91
–
6.76(m,3h),6.63(s,1h),6.56(d,j=8.3hz,1h),6.27(d,j=51.4hz,3h),4.35(d,j=4.7hz,2h),4.20
–
4.04(m,2h),3.85(s,3h),3.59
–
3.43(m,3h),3.33
–
3.20(m,3h),3.05
–
2.85(m,3h),2.62(dd,j=16.7,3.3hz,1h),2.40(s,3h),1.30
–
1.24(m,1h),1.11(t,j=7.2hz,3h).
[0146]
实施例4
[0147][0148]
步骤a:
[0149]
将6-溴代苯并噻吩4-1(1.10克,5.2毫摩尔)加入至二氧六环(30毫升)和水(6毫升)中,然后依次添加(e)-1-乙氧乙烯基-2-硼酸频那醇酯(1.5克,7.7毫摩尔),1,1'-双二苯基膦二茂铁二氯化钯(190毫克,0.26毫摩尔),碳酸钾(2.10克,15.2毫摩尔),之后将混合物氩气置换三次,体系升温至80℃,氩气保护下80℃反应3小时。反应完全后将混合物冷却至室温,用水(30毫升)稀释并用乙酸乙酯(30毫升
×
3)萃取。有机层合并后用无水硫酸钠干燥,过滤并浓缩得到残留物,残留物过硅胶柱(洗脱剂:石油醚/乙酸乙酯=100/1-20/1)纯化得到目标化合物(e)-6-(2-乙氧基乙烯基)苯并[b]噻吩4-2(900毫克),为淡黄色固体。
[0150]
步骤b:
[0151]
将中间体4-2(900毫克,4.41毫摩尔)溶于5毫升四氢呋喃溶液中,并在0℃下加入1.5毫升浓盐酸,之后所得混合物在0℃下搅拌4小时,反应完全后用水(30毫升)稀释再用乙酸乙酯(30毫升
×
3)萃取,有机层合并后用盐水(30毫升)洗涤,用无水硫酸钠干燥,过滤并浓缩,2-(苯并[b]噻吩-6-基)乙醛粗品4-3(700毫克),为淡黄色油状物,粗品直接用于下一步。
[0152]
步骤c:
[0153]
将4-3(700毫克)溶于甲醇(5毫升)中,并在0℃下添加硼氢化钠(151毫克,3.98毫摩尔),之后混合物在室温下搅拌1小时。反应完全后用稀盐酸调反应液ph=5,然后用乙酸
乙酯(50毫升
×
2)萃取。合并后有机层,用饱和食盐水(50毫升)洗涤,再用无水硫酸钠干燥,过滤和浓缩得到残留物。残留物过硅胶柱柱纯化(洗脱剂:石油醚/乙酸乙酯=20/1-2/1)得到目标化合物2-(苯并[b]噻吩-6-基)乙烷-1-醇4-4(290毫克),为淡黄色油状物。
[0154]1h nmr(400mhz,cdcl3)δ7.75(t,j=6.6hz,2h),7.39(d,j=5.6hz,1h),7.30(t,j=2.8hz,1h),7.22
–
7.25(m,1h),3.90(t,j=6.6hz,2h),2.96(t,j=6.4hz,2h).
[0155]
步骤d:
[0156]
将化合物1-(4-氯-2-甲基苯基)-n-乙基-7-羟基-6-甲氧基-3,4-二氢异喹啉-2(1h)-甲酰胺制备例1(80毫克,0.21毫摩尔)加入至5毫升甲苯中,再依次添加三苯基膦(73毫克,0.28毫摩尔),4-4(38.0毫克,0.21毫摩尔),之后将混合物用氩气换气三次。将所得的混合物在120℃下加热10分钟,然后逐滴添加偶氮二甲酸二异丙酯(0.05毫升,0.28毫摩尔)。滴毕,反应混合物在120℃下再搅拌3小时。反应完全后将所得混合物冷却至室温,然后加入10毫升水稀释,再用乙酸乙酯(30毫升
×
2)萃取体系。合并后有机层用20毫升饱和食盐水洗涤,再用无水硫酸钠干燥,有机相过滤浓缩的残留物,残留物通过制备板(二氯甲烷/甲醇=42/1)纯化得到实施例4(25.75毫克),为白色固体。
[0157]
ms(esi):m/z 535.3[m+h]
+
.
[0158]
h nmr(400mhz,dmso-d6)δ7.86(s,1h),7.77(d,j=8.0hz,1h),7.67(d,j=
[0159]
5.2hz,1h),7.39(d,j=5.6hz,1h),7.29
–
7.27(m,1h),7.24(d,j=2.4hz,1h),7.09
–
7.06(m,1h),6.76(s,1h),6.66(t,j=5.2hz,1h),6.53(t,j=4.2hz,2h),6.48(s,1h),4.11
–
4.07(m,1h),4.02
–
3.97(m,1h),3.75(s,3h),3.72
–
3.68(m,1h),3.10
–
3.03(m,4h),2.92
–
2.79(m,2h),2.67
–
2.56(m,1h),2.33(s,3h),1.00(t,j=7.2hz,3h).
[0160]
实施例5
[0161]
[0162][0163]
步骤a:
[0164]
室温下向1,2-二溴乙烷(7.55毫升,87.3毫摩尔),碳酸钾(24.1克,175毫摩尔)和100毫升乙腈的混合液中加入2,5-二溴苯酚5-1(11.0克,43.6毫摩尔)。将反应体系在80℃下搅拌18小时。反应毕混合液冷却至室温,加入200毫升水,再用乙酸乙酯(100毫升*2次)萃取。有机层合并后用饱和食盐水(100毫升)洗涤,再用无水硫酸钠干燥,过滤并减压浓缩得残留物,残留物用硅胶柱纯化(洗脱剂:石油醚/乙酸乙酯=100/1-50/1)得到1,4-二溴-2(2溴乙氧基)苯5-2(4.00克),为无色油状物。
[0165]1h nmr(400mhz,dmso-d6)δ7.54(d,j=8.4hz,1h),7.35(d,j=2.1hz,1h),7.12(dd,j=8.4,2.1hz,1h),4.48
–
4.43(m,2h),3.84
–
3.79(m,2h).
[0166]
步骤b:
[0167]
将正丁基锂(7.31毫升,11.7毫摩尔,1.6m/己烷)缓慢添加到氮气保护-70℃下四氢呋喃(5毫升)和1,4-二溴-2(2溴乙氧基)苯5-2(4.20克,11.7毫摩尔)的溶液中。滴毕,反应液在-70℃下搅拌1小时,然后加入5毫升乙酸淬灭反应。再加入50毫升水稀释反应液,用乙酸乙酯(50毫升*2次)萃取。有机层合并后用50毫升饱和食盐水洗涤,再用无水硫酸钠干燥,有机相过滤并减压浓缩残留物,残留物用硅胶柱纯化(洗脱剂:石油醚/乙酸乙酯=1/0-100/1)得6-溴-2,3-二氢苯并呋喃5-3(1.20g),为白色固体。
[0168]1h nmr(400mhz,cdcl3)δ7.03(d,j=7.8hz,1h),6.98
–
6.92(m,2h),4.58(t,j=8.7hz,2h),3.15(t,j=8.5hz,2h).
[0169]
步骤c:
[0170]
将6-溴-2,3-二氢苯并呋喃6-3(1.15克,5.84毫摩尔),(e)-1-乙氧乙烯基-2-硼酸频那醇酯(1.73克,8.76毫摩尔)、碳酸钾(2.42克,17.5毫摩尔)和1,1'-双(二苯基膦)二茂铁二氯化钯(427毫克,0.58毫摩尔)加入至20毫升二氧六环和5毫升水的混合液中,反应体系氩气置换三次再升温至80℃,并在此温度下搅拌2小时。加入50毫升水50毫升稀释反应液,再用乙酸乙酯(50毫升*2次)萃取。有机层合并后用50毫升饱和食盐水洗涤,再用无水硫酸钠干燥,有机相过滤并减压浓缩得残留物,残留物用硅胶柱纯化(洗脱剂:石油醚/乙酸乙酯=20/1-1/1)得5-4(1.00克),为淡黄色固体。
[0171]
ms(esi):m/z 191.2[m+h]
+
.
[0172]
步骤d:
[0173]
0℃下,向(e)-6-(2-乙氧基乙烯基)-2,3-二氢苯并呋喃5-4(200毫克,1.06毫摩尔)和5毫升四氢呋喃的溶液中加入1.1毫升浓盐酸,体系在0℃下搅拌3小时,然后加入20毫升水,再用乙酸乙酯(20毫升*2次)萃取。有机层合并,用20毫升饱和食盐水洗涤,再用无水硫酸钠干燥,过滤并减压浓缩得到2-(2,3-二氢苯并呋喃-6-基)乙醛粗品5-5(554毫克),为黄色油状物,直接用于下一步反应。
[0174]
步骤e:
[0175]
0℃下,向中间体5-5(554毫克,3.42毫摩尔)和甲醇(10毫升)的溶液中加入硼氢化钠(51毫克,1.06毫摩尔)。混合物在室温下搅拌1小时。用1当量(1n))稀盐酸调体系ph=5,然后用乙酸乙酯(50毫升*2)萃取。有机层合并后用50毫升饱和食盐水洗涤,再用无水硫酸钠干燥,有机相过滤并减压浓缩得残留物,残留物用硅胶柱纯化(洗脱剂:石油醚/乙酸乙酯=10/1-3/1)得到5-6(230毫克),为浅棕色油状物。
[0176]1h nmr(400mhz,cdcl3)δ7.12(d,j=7.6hz,1h),6.70
–
6.72(m,1h),6.67(s,1h),4.56(t,j=8.8hz,2h),3.83
–
3.84(m,2h),3.18(t,j=8.6hz,2h),2.82(t,j=6.4hz,2h).
[0177]
步骤f:
[0178]
实施例5的制备按照类似实施例4的方法得到。用5-6(43.9毫克,0.27毫摩尔)和制备例1(100毫克)得到白色固体实施例5(24.37毫克)。
[0179]
ms(esi):m/z 521.3[m+h]
+
.
[0180]1h nmr(400mhz,dmso-d6)δ7.24(d,j=2.0hz,1h),7.06
–
7.09(m,2h),6.76(s,1h),6.65
–
6.69(m,3h),6.48
–
6.53(m,3h),4.47(t,j=8.6hz,2h),3.98
–
4.00(m,1h),3.86
–
3.88(m,1h),3.76(s,3h),3.68
–
3.75(m,1h),3.05
–
3.11(m,4h),2.83
–
2.92(m,4h),2.56
–
2.80(m,1h),2.39(s,3h),1.00(t,j=7.0hz,3h).
[0181]
实施例6
[0182]
[0183][0184]
步骤a:
[0185]
氩气保护下将6-1(1.4克,7.1毫摩尔),(e)-1-乙氧乙烯基-2-硼酸频那醇酯(2.10克10.6毫摩尔),1,1'-双(二苯基膦)二茂铁二氯化钯(260毫克,0.36毫摩尔),碳酸钾(2.00克,14.5毫摩尔),1,4-二氧六环(30毫升)和水(6毫升)的混合物加热至80℃搅拌3小时。将反应液冷却至室温后经过滤后加水(20毫升)并用乙酸乙酯(3x 30毫升)萃取。合并后的有机相经干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯20/1~10/1),得到浅黄色固体6-2(1.10克)。
[0186]1h nmr(400mhz,cdcl3)δ8.01(s,1h),7.64(d,j=8.4hz,1h),7.40(s,1h),7.27-7.23(m,1h),7.03(d,j=12.8hz,1h),5.95(d,j=13.2hz,1h),3.93(q,j=7.1hz,2h),1.36(t,j=7.0hz,3h).
[0187]
步骤b:
[0188]
将6-2(250毫克,1.321毫摩尔)的甲酸(3.5毫升)溶液于室温下搅拌3小时。所得混合物加饱和碳酸氢钠水溶液(30毫升)后用乙酸乙酯(2
×
30毫升)萃取。合并后的有机相经饱和食盐水(20毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,得到粗品黄色油状物6-3(213mg)。可以直接用于下一步反应。
[0189]
步骤c:
[0190]
0℃下向搅拌着的粗品6-3(213mg,1.321毫摩尔)的甲醇(10ml)溶液中加入硼氢化钠(50mg,1.321毫摩尔)。反应液随后同温下搅拌1小时。向反应液中加入稀盐酸(1.0m)淬灭反应,直至ph=5。所得混合物用乙酸乙酯(2
×
50毫升)萃取。合并后的有机相经饱和食盐水(50毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯10/1~1/1),得到6-4(60毫克,棕色油状物)。
[0191]1h nmr(400mhz,cdcl3)δ8.06(s,1h),7.72(d,j=8.0hz,1h),7.48(d,j=0.4hz,
1h),7.25(dd,j=8.4,1.2hz,1h),3.93(t,j=6.4hz,2h),3.02(t,j=6.4hz,2h).
[0192]
步骤d:
[0193]
室温下将制备例1(168毫克0.448毫摩尔),三苯基膦(126mg,0.4785毫摩尔),6-4(60mg,0.368毫摩尔)的甲苯(5ml)溶液氩气抽换气三次,氩气保护下加热至120℃搅拌10分钟。随后逐滴加入偶氮二甲酸二异丙酯(0.08ml,0.4785毫摩尔)。反应液随后120℃并且氩气保护下搅拌3小时。反应液冷却至室温后加水(10ml),所得混合物用乙酸乙酯(2
×
30毫升)萃取。合并后的有机相经饱和食盐水(20毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯10/1~1/2),再经薄层制备色谱分离(展开剂:二氯甲烷/甲醇=40/1),得到白色固体实施例6(35.23毫克)。
[0194]1h nmr(400mhz,dmso-d6)δ8.66(s,1h),7.69-7.66(m,2h),7.30(dd,j=8.2,1.4hz,1h),7.24(d,j=2.4hz,1h),7.07(dd,j=8.4,2.4hz,1h),6.76(s,1h),6.66(t,j=5.4hz,1h),6.53-6.48(m,3h),4.11-4.08(m,1h),4.07-3.97(m,1h),3.75(s,3h),3.72-3.68(m,1h),3.12-3.04(m,4h),2.95-2.75(m,2h),2.56-2.55(m,1h),2.39(s,3h),1.00(t,j=7.2hz,3h).
[0195]
lc-ms(esi):m/z 520.2[m+h]
+
.
[0196]
实施例7
[0197]
[0198]
步骤a:
[0199]
将7-1(2.3克,10.74毫摩尔),乙烯三氟硼酸钾(2.16克,16.12毫摩尔),双(三苯基膦)二氯化钯(754毫克,1.07毫摩尔),碳酸钾(4.45克,32.2毫摩尔),1,4-二氧六环(100毫升)和水(20毫升)的混合物加热至90℃并且氩气保护下搅拌5小时。将反应液冷却至室温后加水(100毫升),所得混合物用乙酸乙酯(3
×
100毫升)萃取。合并后的有机相经饱和食盐水(100毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯=20/1),得到7-2(1.30克),为无色油状物。
[0200]
lc-ms(esi):m/z 162.0[m+h]
+
.
[0201]
步骤b:
[0202]
室温并且氩气保护下向搅拌着的7-2(1.3克,8.06毫摩尔)的四氢呋喃(100毫升)溶液中逐滴加入9-bbn的四氢呋喃溶液(0.5摩尔浓度,24.2毫升,12.1毫摩尔)。反应液随后加热至80℃并且氩气保护下搅拌4小时。将反应液冷却至室温,搅拌下逐滴加入双氧水(30%水溶液,4.57克,40.32毫摩尔)。所得混合物加热至80℃并且搅拌1小时。反应液冷却至室温后加乙酸乙酯(2x100毫升)萃取。合并后的有机相经水(2x 100毫升)洗涤,亚硫酸钠水溶液(50毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯=2/1),得到7-3(620毫克),为浅黄色油状物。
[0203]
lc-ms(esi):m/z 180.1[m+h]
+
.
[0204]
步骤c:
[0205]
室温下将7-3(150毫克,0.837毫摩尔),制备例1(313毫克,0.835毫摩尔)和三苯基膦(263毫克,1.00毫摩尔)的干燥四氢呋喃(10毫升)溶液氩气抽换气三次,氩气保护下冷却至0℃并且搅拌下逐滴加入偶氮二甲酸二异丙酯(0.199毫升,1.00毫摩尔)的四氢呋喃(3毫升)溶液。反应液随后恢复至室温并且氩气保护下搅拌2小时。将反应液浓缩,所得残渣经高效液相制备色谱分离(waters 2767/2545/2489/qda,inertsil ods-3 10um 20*250nm,流动相a:0.1%甲酸水溶液,流动相b:乙腈,流速:20毫升/分钟:柱温:室温),得到实施例7(27.4毫克),为白色固体。
[0206]
lc-ms(esi):m/z 536.3[m+h]
+
.
[0207]1h nmr(400mhz,dmso-d6)δ9.35(s,1h),8.15-7.97(m,2h),7.45-7.37(m,1h),7.24(d,j=2.1hz,1h),7.07(dd,j=8.3,2.2hz,1h),6.76(s,1h),6.67(t,j=5.4hz,1h),6.58-6.42(m,3h),4.14-4.08(m,1h),4.05-3.98(m,1h),3.74(s,3h),3.73-3.65(m,1h),3.16-3.05(m,4h),2.95-2.70(m,2h),2.58-2.53(m,1h),2.38(s,3h),1.00(t,j=7.1hz,3h).
[0208]
实施例8
[0209]
[0210]
实施例8的制备按照类似实施例7的方法得到。用原料2-(苯并【d-】噻唑-6-)基)乙醇-1(200毫克,1.116毫摩尔)和制备例1(349毫克,0.931毫摩尔)反应得到白色固体实施例8(151.26毫克)。
[0211]
lc-ms(esi):m/z 536.1[m+h]
+
.
[0212]1h nmr(400mhz,dmso-d6)δ9.32(s,1h),8.03(d,j=1.2hz,1h),7.98(d,j=8.4hz,1h),7.45(dd,j=8.4,1.6hz,1h),7.24(d,j=2.0hz,1h),7.08(dd,j=8.2,2.2hz,1h),6.77(s,1h),6.67(t,j=5.4hz,1h),6.55-6.50(m,2h),6.49(s,1h),4.15-4.08(m,1h),4.04-3.96(m,1h),3.75(s,3h),3.74-3.67(m,1h),3.14-3.03(m,4h),2.97-2.87(m,1h),2.86-2.75(m,1h),2.58-2.52(m,1h),2.39(s,3h),1.00(t,j=7.1hz,3h).
[0213]
实施例9
[0214][0215]
步骤a:
[0216]
室温并且氩气保护下向9-1(8.00克,40.6毫摩尔),1,4-二氧六环(150毫升)和水(50毫升)的混合物中加入乙烯三氟硼酸钾(8.16克,60.9毫摩尔),1,1'-双(二苯基膦)二茂铁二氯化钯(1.48克,2.03毫摩尔)和碳酸铯(39.7克,122毫摩尔)。反应液随后加热至100℃并且氩气保护下搅拌16小时。反应液冷却至室温后抽滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯=4/1),得到9-2(4.30克),为黄色固体。
[0217]
lc-ms(esi):m/z 145.1[m+h]
+
.
[0218]
步骤b:
[0219]
室温下向搅拌着的9-2(1.00克,6.94毫摩尔)的四氢呋喃(10毫升)溶液中加入二碳酸二叔丁酯(4.45毫升,20.8毫摩尔)和4-二甲氨基吡啶(0.42克,3.47毫摩尔。反应液随后室温下搅拌3小时。反应液加水(100毫升)稀释后用二氯甲烷(2
×
100毫升)萃取。合并后的有机相经饱和食盐水(100毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯=100/1~80/1),得到9-3(1.10克),为黄色固体。
[0220]
lc-ms(esi):m/z 189.1[m-t-bu+2h]
+
.
[0221]
步骤c:
[0222]
0℃并且氩气保护下向搅拌着的硼烷四氢呋喃溶液(1.0m,4.91毫升,4.91毫摩尔)和干燥四氢呋喃(20毫升)混合物中逐滴加入9-3(800毫克,3.28毫摩尔)的四氢呋喃(30毫升)溶液。反应液随后恢复至室温并且氩气保护下搅拌过夜。逐滴加入氢氧化钠水溶液(3.0m,1.64毫升,4.92毫摩尔)和双氧水(30%水溶液,0.493毫升,4.91毫摩尔)。所得混合物加热至80℃并且搅拌5小时。反应液冷却至室温后加水(40毫升)稀释,所得混合物用二氯甲烷(2
×
100毫升)萃取。合并后的有机相经饱和食盐水(30毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:二氯甲烷/甲醇=40/1),得到9-4(500mg),为黄色固体。
[0223]
lc-ms(esi):m/z 263.0[m+h]
+
.
[0224]
步骤d:
[0225]
室温下将9-4(200毫克,0.762毫摩尔),30-3(239毫克,0.635毫摩尔)和三苯基膦(200毫克,0.762毫摩尔)的干燥四氢呋喃(10毫升)溶液氩气抽换气三次,氩气保护下冷却至0℃并且搅拌下逐滴加入偶氮二甲酸二异丙酯(0.151毫升,0.762毫摩尔)的四氢呋喃(10毫升)溶液。反应液随后恢复至室温并且氩气保护下搅拌3小时。将反应液浓缩,所得残渣加水(50毫升)稀释后用二氯甲烷(2
×
100毫升)萃取。合并后的有机相经饱和食盐水(100毫升)洗涤,无水硫酸钠干燥并过滤。滤液浓缩,所得残渣经硅胶柱层析分离(洗脱剂:二氯甲烷/甲醇=50/1~10/1),得到9-5(210毫升),为黄色固体。
[0226]
lc-ms(esi):m/z 619.2[m+h]
+
.
[0227]
步骤e:
[0228]
将9-5(150毫克,0.242毫摩尔)和氯化氢/1,4-二氧六环溶液(4.0m,10毫升,40毫摩尔)的混合物在室温下搅拌4小时。反应液浓缩,所得残渣经高效液相制备色谱分离(waters 2767/2545/2489/qda,inertsil ods-3 10um 20*250nm,流动相a:0.1%甲酸溶液,流动相b:ch3cn,流速:20毫升/分钟:柱温:室温),得到实施例9(33.16毫克),为白色固体。
[0229]
lc-ms(esi):m/z 519.3[m+h]
+
.
[0230]1h nmr(400 mhz,dmso-d6)δ12.93(s,1h),7.96(s,1h),7.58(s,1h),7.43(d,j=8.5 hz,1h),7.29-7.17(m,2h),7.08(dd,j=8.3,2.1 hz,1h),6.76(s,1h),6.66(t,j=5.3 hz,1h),6.55-6.47(m,3h),4.10-4.04(m,1h),3.96-3.92(m,1h),3.75(s,3h)3.74-3.66(m,1h),3.13-2.98(m,4h),2.96-2.72(m,2h),2.56-2.51(m,1h),2.39(s,3h),1.00(t,j=7.1 hz,3h).
[0231]
实施例10
[0232][0233]
实施10的制备按照类似实施例9的方法得到。用2-(苯并【d-】吡唑-6-)基)乙醇-1(100毫克,0.381毫摩尔)和制备例1(142毫克,0.379毫摩尔)反应得到白色固体中间体(30毫克)。
[0234]
将中间体(30毫克,0.048毫摩尔)和氯化氢/1,4-二氧六环溶液(4.0 m,3毫升,12毫摩尔)的混合物在室温下搅拌2小时。反应液浓缩,所得残渣经高效液相制备色谱分离(流动相含碳酸氢铵),得到实施例10(6.67 mg),为白色固体。
[0235]
lc-ms(esi):m/z 519.3[m+h]
+
.
[0236]1h nmr(400 mhz,dmso-d6)δ12.91(s,1h),7.98(s,1h),7.63(d,j=8.0 hz,1h),7.40(s,1h),7.24(d,j=2.0 hz,1h),7.09-7.00(m,2h),6.76(s,1h),6.67(t,j=5.2 hz,1h),6.54-6.48(m,3h),4.11-4.04(m,1h),4.00-3.92(m,1h),3.74(s,3h),3.74-3.69(m,1h),3.10-3.04(m,4h),2.96-2.65(m,2h),2.60-2.52(m,1h),2.39(s,3h),1.00(t,j=7.1 hz,3h).
[0237]
实施例11
[0238][0239]
实施例11的制备按照类似实施例5的方法得到。用原料5-(2-羟乙基)-2,3-二氢苯并呋喃(130毫克,0.79毫摩尔)和制备例1(296毫克)反应得到白色固体实施例11(51.66毫克)。
[0240]
ms(esi):m/z 521.2[m+h]
+
[0241]1h nmr(400mhz,dmso-d6)δ7.25(d,j=2.1hz,1h),7.10
–
7.06(m,2h),6.93(d,j=8.1hz,1h),6.76(s,1h),6.67(t,j=5.4hz,1h),6.62(d,j=8.1hz,1h),6.53(d,j=8.3hz,1h),6.49(d,j=3.0hz,2h),4.46(t,j=8.7hz,2h),3.97(dd,j=16.4,7.5hz,1h),3.85(dd,j=16.8,7.3hz,1h),3.75(s,3h),3.72
–
3.66(m,1h),3.10
–
3.00(m,4h),2.96
–
2.74(m,4h),2.56(d,j=3.2hz,1h),2.39(s,3h),1.00(t,j=7.1hz,3h).
[0242]
实施例12
[0243][0244]
实施例12的制备按照类似实施例5的方法得到。用原料2-(苯并[d][1,3]二氧醇-5-基]乙烷-1-乙烷-1-1-醇(300mg,1.81mmol)和制备例1(564毫克,1.51毫摩尔)反应得到白色固体实施例12(206.5毫克)。
[0245]
ms(esi):m/z 523.2[m+h]
+
[0246]1h nmr(400mhz,dmso-d6))δ7.24(d,j=1.2hz,1h),7.10
–
7.05(m,1h),6.79(s,1h),6.78
–
6.75(m,2h),6.74
–
6.65(m,2h),6.52
–
6.48(m,3h),5.93(s,2h),4.01
–
3.95(m,1h),3.89
–
3.85(m,1h),3.88
–
3.74(m,4h),3.15
–
3.00(m,2h),2.90
–
2.75(m,4h),2.39(s,3h),1.25(s,1h),1.00(t,j=7.2hz,3h).
[0247]
实施例13
[0248][0249]
步骤a:
[0250]
在干燥的250毫升三口烧瓶中依次加入化合物2-(2,3-二氢苯并[b][1,4]二恶英-6-基)乙酸13-1(1.00克,5.15毫摩尔)和无水四氢呋喃溶液(30毫升),0度下滴加硼烷(8.0毫升,8.0毫摩尔,1.0m)的四氢呋喃(30毫升)溶液,然后升至室温,搅拌反应4小时。薄板层析(tlc)监测反应结束后,反应液减压浓缩,加入冰水(80毫升),乙酸乙酯(60毫升
×
2)萃取。有机层合并,用盐水(30毫升)洗涤,无水硫酸钠干燥,过滤并浓缩以得到13-2(780毫克,黄色油状)。
[0251]
lc-ms(esi):m/z 163.0[m-oh]
+
.
[0252]
步骤b:
[0253]
实施例13的制备按照类似实施例6的方法得到。用2-(2,3-二氢苯并[b][1,4]二恶英-6-基乙烷-1-乙烷-1-醇(200m毫克,1.11毫摩尔)13-2和制备例1(349毫克,0.93毫摩尔)反应得到灰白色固体实施例13(100毫克)。
[0254]
ms(esi):m/z 537.2[m+h]
+
[0255]1h nmr(400mhz,cdcl3)δ7.19(d,j=2.1hz,1h),6.99(dd,j=8.2,2.5hz,1h),6.76(d,j=8.2hz,1h),6.72(d,j=2.0hz,1h),6.68
–
6.61(m,3h),6.48(s,1h),6.36(s,1h),4.22(s,4h),4.06
–
3.90(m,2h),3.86(s,3h),3.57
–
3.47(m,1h),3.33
–
[0256]
3.22(m,3h),3.04
–
2.89(m,3h),2.64(d,j=15.4hz,1h),2.48(s,3h),1.63(s,1h),1.12(t,j=7.2hz,3h).
[0257]
实施例14
[0258][0259]
实施例14的制备按照类似实施例4的方法得到。用原料2-(苯并[b]噻吩-5-基]乙烷-1-醇(200毫克,1.12毫摩尔)和制备例1(350毫克)得到白色固体实施例14(62.55毫克)。
[0260]
ms(esi):m/z 535.2[m+h]
+
.
[0261]1h nmr(400mhz,dmso-d6)δ7.88(d,j=8.2hz,1h),7.76
–
7.70(m,2h),7.37(d,j=5.5hz,1h),7.27(d,j=8.4hz,1h),7.24(d,j=1.9hz,1h),7.09
–
7.06(m,1h),6.76(s,1h),6.67(t,j=5.2hz,1h),6.52(t,j=4.1hz,2h),6.48(s,1h),4.11
–
4.06(m,1h),4.02
–
3.95(m,1h),3.75(s,3h),3.70(s,1h),3.06(t,j=6.4hz,4h),2.96
–
2.75(m,2h),2.56(s,1h),2.38(s,3h),1.00(t,j=7.1hz,3h).
[0262]
实施例15
[0263][0264]
实施例15的制备按照类似实施例14的方法得到。用2-(苯并呋喃-5-基)乙烷-1-1-醇(150mg,0.926mmol)和制备例1(346毫克,0.926毫摩尔)反应得到白色固体实施例15(9.27毫克)。
[0265]
ms(esi):m/z 519.2[m+h]
+
[0266]1h nmr(400mhz,dmso-d6))δ7.93(d,j=2.4hz,1h),7.51(s,1h),7.46(d,j=8.4hz,1h),7.24(d,j=2.4hz,1h),7.20(dd,j=8.4,1.6hz,1h),7.07(dd,j=8.0,1.5hz,
1h),6.87(dd,j=2.4,1.2hz,1h),6.76(s,1h),6.67(t,j=5.4hz,1h),6.54
–
6.51(m,2h),6.48(s,1h),4.10
–
4.04(m,1h),3.98
–
3.91(m,1h),3.75(s,3h),3.74
–
3.68(m,1h),3.12
–
3.00(m,4h),2.96
–
2.74(m,2h),2.56
–
2.51(m,1h),2.38(s,3h),1.00(t,j=7.2hz,3h).
[0267]
测试例1:细胞增殖抑制实验
[0268]
选取商业化来源的4株肿瘤细胞系panc-1(g12d)、h358(g12c)、a549(g12s)、hct116(g13d)开展细胞增殖抑制实验,分别培养在含10%胎牛血清的dmem、dmem、f12k、mccoy's 5a、rpmi-1640和emem培养基(gibco,thermofisher)中,放置于37℃、5%的co2培养箱中孵育。细胞均呈贴壁状态生长,在倒置显微镜下观察生长状况,待细胞数量适量时传代培养。
[0269]
取对数生长期的panc-1、h358、a549、hct116细胞,以合适的细胞密度接种于96孔细胞培养板中(corning),在含5%co2的细胞培养箱中37℃培养24h后,每孔分别加入10μl待测化合物或阳性药物。同时设置阳性对照组(100%抑制孔)及阴性对照组(0%抑制孔),药物组每浓度重复2孔,阳性对照组和阴性对照组重复6孔,培养箱中继续培养5天后,接后续alamarblue测试操作;
[0270]
alamarblue测试操作:每孔加10μl alamarblue试剂(thermofisher)孵育1-4h,振荡1-2min,md酶标仪ex:560nm,em:590nm波长测得荧光值,记录结果,通过计算药物对细胞抑制率(%)=(a
0%抑制
-a
药物
)/(a
0%抑制
-a
100%抑制
)
×
100%,再利用matilab软件采用非线性回归的方法(常采用四参数拟合曲线方程,4-parameter logistic model))作图得到药物剂量反应曲线,从而获得化合物的ic50值及其他相关参数。测试化合物对6株商业化肿瘤细胞系(panc-1、h358、a549和hct116)的增殖抑制活性(ic
50
,μm)结果如下表1所示。
[0271]
化合物生物活性数据
[0272]
表1:细胞增殖抑制(ic
50
:μm)
[0273]
[0274][0275]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1