新型噻唑类化合物及其制备方法和用途与流程
1.本发明涉及一种新型噻唑类化合物及其前药、药学上可接受的盐、晶体和它们的药物组合物、以及它们的制备方法和用途。
背景技术:
2.病毒性感染一直是人类健康的一大挑战。慢性乙型肝炎病毒(hbv)感染是在全球范围内导致严重肝脏疾病的常见原因。中国每年新发乙肝病毒感染人数高达100万以上,总感染人数达9000万。根据世界卫生组织(who)估计,全球有大约2.57亿hbv感染患者。每年约超过65万人死于hbv感染相关终末期肝病,包括肝功能衰竭、肝硬化和肝细胞癌(hcc)。目前,疫苗、核苷(na)或核苷酸(nuc)类药物能够有效地降低新感染率,而且对于坚持长期病毒抑制疗法的hbv患者来说,能够延缓肝病的进展。乙肝表面抗原(hbsag)阴转与肝脏功能改善、组织病理改善以及长期预后改善相关,是目前国内外最新慢性乙肝防治指南推荐的理想治疗目标,即乙型肝炎的功能性治愈或称为临床治愈的目标。然而直接抗病毒药物(daa)[如核苷(酸)类似物(na)]或免疫调节剂[如聚乙二醇化干扰素α(peg-ifn)]单独使用实现临床治愈的作用有限。理论上,na和peg-ifn针对hbv发挥不同的抗病毒作用,合理联用能够产生协同和互补的效应,但仍然治愈率很低。目前一些具有新的作用机理的药物开始进入临床试验,如免疫检查点抑制剂(抗pd-1或抗ctla-4疗法),衣壳组装抑制剂(at-130,nvr-3778,jnj6379等),rnai疗法(jnj3989或aro-hbv)和细胞凋亡蛋白抑制剂(iap,inhibitors of apoptosi s proteins)抑制剂(如apg-1387)等(fanning et al.,volume 18,pages827
–
844(2019))。这些都不能实现乙肝功能性治愈的目标。因此需要发现更有效的乙肝治疗药物,提高其功能性治愈率。
[0003]
covid-19自2019年底在全球爆发到2021年3月17日止,全球感染人数已达到1.17亿,死亡人数超过260万,还没有明显结束象征。虽然有几款疫苗已经授予紧急使用权,但还没有有效药物正式批准上市。急需发现疗效确切的治疗药物,解决目前covid-19引起的健康危机。
[0004]
脂肪肝的发病率不断升高,伴随着由其引起的肝纤维化发病率的上升,但是没有有效的药物治疗。因此,急需发现有效治疗药物。
[0005]
近年来的研究发现,硝唑尼特具有广谱抗病毒作用(j.genetic engineering and biotechology 2020,18,1),能有效抑制包括乙肝病毒(hbv),丙肝病毒(hcv),艾滋病病毒(hiv)和新冠状病毒(covid-19),流感病毒(influenza a/b)等病毒的复制。化合物1对乙肝病毒的多种蛋白,包括hbsag,hbeag,hbcag,hbv r.i.,hbv rna,cccdna和hbv的变异株都有显著的抑制作用(antiviral res.2008,77,56)。硝唑尼特能有效杀死多药耐药革兰氏阴性菌(cid 2013,56,1685),显著抑制新冠病毒(j.infect.dev.ctries.2020,14,982),治疗呼吸道感染也有效(doi:10.1093/cid/ciz100),能有效抑制流感病毒(antimicrob.agents chemother.2015,59,1061),具有广谱抗病毒包括抗hiv活性(j.genetic engineer.biotech.2020,18,35),对肺结核有效(j.med.chem.2009,52,5789),对治疗脂肪
肝炎引起的纤维化有效(us pat.2018/0092885a1),对多种癌症也有效(mutation research/fundamental and molecular mechanisms of mutagenesis,volume 768,october 2014,pages 16-21)。中国专利cn201610329100.8还披露了化合物1用于阿兹海默症和帕金森病的用途。
[0006]
硝唑尼特及其代谢活性药物替唑尼特(tizoxanide,2)水溶性很差,口服只有31.5%吸收,66.2%从大便排出(int.j.clin.pharmacol.ther.2000,38,387)。这对于治疗胃肠道疾病是可行的。但是,用于系统性治疗病毒感染,药物必须被有效吸收才能发挥药物作用。制备带有碱性基团的前药可以成盐,提高其水溶性,可能改善其生物利用度。stachulski等报道,氨基酸酯盐酸盐(rm5061)的生物利用度从化合物1的2.8%提高到20%,(eur.j.med.chem.2017,126,154)。
[0007]
据报道,化合物1的活性成分极易被糖甙化生成没有生物活性的化合物5(tzg,tizoxanide glucuronide),血浆中的药物浓度有时难以达到有效治疗浓度。化合物2的任何形式的前药都不能改变其羟基被糖甙而失去生物活性的特性。
[0008]
据文献报道,2-甲基取代化合物(2-me-2)抑制丙肝病毒的活性明显低于化合物1,ec
90
降低了8倍(j.med.chem.2011,54,8670)。2-甲基取代化合物(2-me-2)抗乙肝病毒的活性也明显降低,ec
50
降低了4倍(j.med.chem.2011,54,4119)。
[0009]
技术实现要素:
[0010]
本发明的目标之一是提供一种生物活性更强的新型噻唑类化合物,本发明的另一个目标是提供一种显著减少活性成分的糖甙化,或显著提高药物暴露量的新型噻唑类化合物。
[0011]
发明人研究发现,rm5061并不能有效提高药物的暴露量。发明人设想将化合物1或2的邻位的h由不同的取代基取代,以阻止其羟基的糖甙化,有效地提高药物的暴露量。发明人惊奇地发现邻位被乙基(i-1a-8)和异丙基(i-1a-12)取代后抗乙肝表面抗原的活性反而
提高了,i-1a-8和i-1a-12的ec
50
分别是化合物2的9.5和2.7倍。化合物i-1a-12在大鼠灌胃后药物暴露量是化合物2的12.5倍。
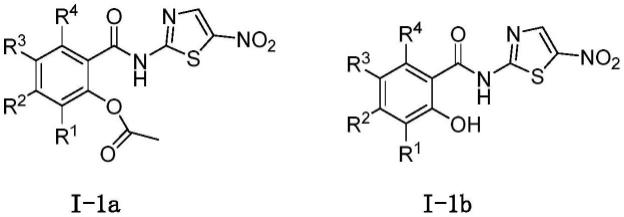
[0012][0013]
在此基础上,提供一种式i所示的化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体,
[0014][0015]
其中:
[0016]
x选自no2、cl、br;
[0017]
r1选自,但不限于:烷基或取代烷基,烯基或取代烯基,炔基或取代炔基,环烷基或取代环烷基,杂环基或取代杂环基,芳基或取代芳基,烷氧基或取代烷氧基,卤素,氰基,硝基,羟基,-ocor’,-ocoor’,-oconhr’,氨基,-nhcor’,-nhcoor’,-nhconhr’,改性烷基,氨基酸酯基
[0018]
r2、r3、r4各自独立地,但不限于选自:氢、烷基或取代烷基,烯基或取代烯基,炔基或取代炔基,环烷基或取代环烷基,杂环基或取代杂环基,芳基或取代芳基,烷氧基或取代烷氧基,卤素,氰基,硝基,羟基,-ocor',-ocoor',-oconhr',氨基,-nhcor',-nhcoor',-nhconhr',改性烷基,氨基酸酯基
[0019]
任选地,r1、r2为烷基时,所述烷基上任意一个或多个氢被氘替代;
[0020]
任选地,r1和r2,或者r2和r3,或者r3和r4形成3-7元饱和或不饱和环;
[0021]
r选自氢、-cor',-coor',-conhr',氨基,-nhcor',-nhcoor',-nhconhr',烷基,改性烷基,或
[0022]
各个r'相互独立地选自,但不限于:氢、烷基或取代烷基,烯基或取代烯基,炔基或取代炔基,环烷基或取代环烷基,杂环基或取代杂环基,芳基或取代芳基;
[0023]
其中中2个r”各自独立地选自:氢、烷基或取代烷基,烯基或取代烯基,炔基或取代炔基,环烷基或取代环烷基,杂环基或取代杂环基,芳基或取代芳基;
[0024]
所述取代烷基、取代烯基、取代炔基、取代环烷基、取代芳基、和取代杂环基中的取代基各自独立地选自,但不限于:1-3个卤素,氰基,氨基,硝基,羟基,烷基,烷氧基,改性烷基,氨基酸酯基;
[0025]
所述氨基酸酯基包括消旋的和光学纯的氨基酸基及其药学上可接受的盐,
[0026]
所述“卤素”选自:f、cl、br或i;
[0027]
所述“烷基”、“烷氧基”中的“烷基”为c
1-12
直链或支链烷基,可选为c
1-8
直链或支链烷基,可选为c
1-5
直链或支链烷基;可选为:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、仲丁基、或正戊基;
[0028]
所述“环烷基”为c
3-8
单环或双环环烷基;
[0029]
所述“烯基”为c
2-c6的烯基;
[0030]
所述“炔基”为c
2-c6的炔基;
[0031]
所述“杂环基”为环上含有1个、2个或3个选自n、o、s的杂原子的3-10元非芳香杂环;
[0032]
所述“芳基”为6-10元芳基;可选为苯基或萘基;
[0033]
所述“改性烷基”为烷基的任意碳原子以及,如果存在,其上的氢原子一起,被选自-o-、-co-、-nh2、-oh、卤素、-cn、-no2中的一种或几种基团置换所得的基团;
[0034]
所述式i化合物不为以下化合物3和4:
[0035][0036]
任选地,任意一个或多个氢被氘替代。
[0037]
可选地,
[0038]
r1选自,但不限于:烷基,卤素,氰基,硝基,或卤代烷氧基;
[0039]
r2选自,但不限于:氢,烷基,或烷氧基;
[0040]
r3选自,但不限于:氢,羟基,烷氧基,卤素,氨基,或-nhcor';
[0041]
r4选自,但不限于:氢,羟基,烷氧基,-nhcor',或氨基;
[0042]
r选自,但不限于:氢、-cor',-coor',-conhr',氨基,-nhcor',-nhcoor',-nhconhr',
[0043]
可选地,
[0044]
各个r'和r”各自独立地选自,但不限于:氢、甲基、乙基、正丙基、异丙基、正丁基、异丁基、苄基、仲丁基、正戊基、环丙基,环丁基,环戊基、环己基、苄基、苯基和取代苯基。
[0045]
可选地,式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体中,所述的式i化合物选自下列化合物i-1:
[0046][0047]
其中:r1、r2、r3、r4和r的定义如上所述。
[0048]
可选地,所述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体中,所述的式i化合物选自化合物i-1a或i-1b:
[0049][0050]
化合物i-1a和i-1b中:r1、r2、r3和r4的定义如上所述。
[0051]
可选地,所述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体中,所述的式i化合物选自化合物i-1a或i-1b:
[0052][0053]
化合物i-1a和i-1b中:
[0054]
r1选自,但不限于:烷基,卤素,氰基,硝基,或卤代烷氧基;
[0055]
r2选自,但不限于:氢,烷基,或烷氧基;
[0056]
r3选自,但不限于:氢,羟基,烷氧基,卤素,氨基,或-nhcor';
[0057]
r4选自,但不限于:氢,羟基,烷氧基,-nhcor',或氨基;
[0058]
可选地,
[0059]
r1选自:甲基,乙基,异丙基,f,br,氰基,硝基,或三氟甲基;
[0060]
r2选自:氢,甲基,或甲氧基;
[0061]
r3选自:氢,羟基,甲氧基,br,氨基,或-nhac;
[0062]
r4选自:氢,羟基,甲氧基,-nhac,或氨基。
[0063]
可选地,所述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体中,所述的式i化合物选自化合物i-1a或i-1b:
[0064][0065]
其中,化合物i-1a、i-1b分别选自下述化合物i-1a-3~i-1a-39、以及i-1b-3~i-1b-39:
[0066]
[0067]
[0068][0069]
可选地,所述药学上可接受的盐包括式i化合物的阴离子盐或阳离子盐;
[0070]
可选地,所述药学上可接受的盐包括式i化合物的金属盐和铵盐;所述金属包括na、k、mg、ca、li、或zn;
[0071]
可选地,所述药学上可接受的盐包括式i化合物与无机酸或者有机酸形成的盐;
[0072]
可选地,所述无机酸包括盐酸、氢溴酸、氢碘酸、硫酸、硝酸、磷酸、或碳酸;
[0073]
可选地,所述有机酸包括甲酸、抗坏血酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、枸橼酸、酒石酸、葡萄糖酸、酒石氢酸、葡萄糖醒酸、碳酸、苦味酸、甲磺酸、乙磺酸、对甲苯磺酸、苯甲酸、苯磺酸、对溴苯磺酸、谷氨酸、水杨酸、或双羟萘酸;
[0074]
可选地,所述药学上可接受的盐为式i化合物的盐酸盐;
[0075]
可选地,所述药学上可接受的盐为在式i化合物的r基团处与酚羟基形成的nh4盐或金属盐,所述金属选自,但不限于,na、k、mg、ca、li、或zn。
[0076]
本发明还提供化合物i-1a-12的晶型a、b、c、d、e、f、h,其中,使用cu-kα辐射,所述晶型a在x射线粉末衍射图谱中2θ值=11.32
°
,12.01
°
,12.76
°
,16.39
°
,18.81
°
,19.61
°
,20.46
°
,22.74
°
,23.58
°
,24.55
°
,25.84
°
,27.15
°
,29.15
°
,35.24
°
的衍射角处有特征峰;
[0077]
使用cu-kα辐射,所述晶型b的x射线粉末衍射图谱中2θ值=7.70
°
,9.32
°
,13.04
°
,15.41
°
,16.31
°
,23.25
°
,25.15
°
,25.57
°
,31.62
°
,39.20
°
,39.60
°
的衍射角处有特征峰;
[0078]
使用cu-kα辐射,所述晶型c的xrpd图谱中2θ值=7.69
°
,13.05
°
,15.43
°
,16.31
°
,23.23
°
,25.57
°
,31.62
°
,39.61
°
的衍射角处有特征峰;
[0079]
使用cu-kα辐射,所述晶型d的x射线粉末衍射图谱中2θ值=10.45
°
,10.68
°
,12.96
°
,16.73
°
,17.97
°
,18.26
°
,23.97
°
,24.45
°
,25.38
°
的衍射角处有特征峰;
[0080]
使用cu-kα辐射,所述晶型e的xrpd谱中2θ值=9.63
°
,10.08
°
,12.40
°
,13.33
°
,23.38
°
,23.89
°
,24.30
°
的衍射角处有特征峰;
[0081]
使用cu-kα辐射,所述晶型f在x射线粉末衍射图谱中2θ值=7.64
°
,9.27
°
,13.00
°
,15.38
°
,16.27
°
,22.14
°
,23.19
°
,24.24
°
,25.53
°
,31.56
°
,39.55
°
的衍射角处有特征峰;
[0082]
使用cu-kα辐射,所述晶型h在x射线粉末衍射图谱中2θ值=10.60
°
,14.04
°
,15.7
°
,18.42
°
,19.70
°
,25.73
°
,26.44
°
的衍射角处有特征峰。
[0083]
本发明另一方面,提供一种上述式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体的制备方法,包括将化合物ii-1与化合物ii-2反应得到式i化合物的步骤;
[0084][0085]
其中,r、r1、r2、r3、r4和x的定义如上所述。
[0086]
本发明另一方面,提供一种药物组合物,包含上述式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体中的一种或多种,以及药学上可接受的载体;
[0087]
可选地,所述药物组合物的剂型包括口服的固体制剂、或液体制剂;
[0088]
可选地,所述固体制剂包括片剂、粉剂、颗粒剂、或胶囊剂;
[0089]
可选地,所述液体制剂包括水或生理盐水或油的悬浮剂、或糖浆。
[0090]
可选地,所述药物组合物还包括除式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体以外的活性化合物。
[0091]
本发明另一方面,提供一种上述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物在制备预防或治疗病毒感染的药物中的用途。
[0092]
本发明另一方面,提供一种上述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物在制备预防或治疗乙型肝炎的药物中的用途。
[0093]
本发明另一方面,提供一种上述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物在制备预防或治疗冠状病毒感染的药物中的用途;
[0094]
可选地,所述冠状病毒包括covid-19、hcov-229e、hcov-oc43、hcov-nl63、hcov-hku1、sars-cov和mers-cov。
[0095]
本发明另一方面,提供一种上述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物在制备预防或治疗肝纤维化的药物中的用途。
[0096]
本发明另一方面,提供一种上述的式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物与其它制剂联用,作为预防或治疗疾病药物中的用途。
[0097]
本发明另一方面,提供一种预防或治疗病毒感染的方法,包括向需要的患者给予预防或治疗有效量的上述式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物。
[0098]
本发明另一方面,提供一种预防或治疗乙型肝炎的方法,包括向需要的患者给予预防或治疗有效量的上述式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物。
[0099]
本发明另一方面,提供一种预防或治疗冠状病毒感染的方法,包括向需要的患者
给予预防或治疗有效量的上述式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;或者所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物。可选地,所述冠状病毒包括covid-19、hcov-229e、hcov-oc43、hcov-nl63、hcov-hku1、sars-cov和mers-cov。
[0100]
本发明另一方面,提供一种预防或治疗肝纤维化的方法,包括向需要的患者给予预防或治疗有效量的上述式i化合物;其前药;所述化合物或其前药的药学上可接受的盐;所述化合物、所述前药或所述盐的晶体;或者上述的药物组合物。
[0101]
本发明任何化合物的氘代物都落在本发明范围内。
[0102]
有益技术效果:
[0103]
一方面,化合物i或其前药或它们药学上可接受的盐或晶体能够显著提高生物活性。
[0104]
另一方面,化合物i或其前药或它们药学上可接受的盐或晶体能有效地减少被糖甙化而失去生物活性,或显著提高有效药物的暴露量,扩大了临床应用。
附图说明
[0105]
图1是晶型a的xrpd谱图
[0106]
图2是晶型b的xrpd谱图
[0107]
图3是晶型c的xrpd谱图
[0108]
图4是晶型d的xrpd谱图
[0109]
图5是晶型e的xrpd谱图
[0110]
图6是晶型f的xrpd谱图
[0111]
图7是晶型h的xrpd谱图
[0112]
图8是晶型h的dsc谱图
[0113]
图9是晶型h的tga谱图
[0114]
图10是晶型h的1h nmr谱图
[0115]
图11是晶型b稳定性研究中的xrpd谱图
[0116]
图12是晶型b稳定性研究中的hplc图
[0117]
图13和图14是晶型h稳定性研究中的xrpd谱图
[0118]
图15和图16是晶型h稳定性研究中的hplc图
[0119]
图17是晶型b的研磨与压片试验中xrpd谱图
[0120]
图18是晶型h的研磨与压片试验中xrpd谱图
[0121]
图19是晶型b的dvs测试结果图
[0122]
图20是晶型b的dvs测试前后的xrpd谱图
[0123]
图21是晶型h的dvs测试结果图
[0124]
图22是晶型h的dvs测试前后的xrpd谱图
[0125]
图23是另一批次晶型h样品的dvs复测结果图
[0126]
图24是另一批次晶型h样品的dvs复测前后的xrpd谱图
[0127]
图25是晶型b的标准曲线
[0128]
图26是晶型b测试完水中溶解度后的滤饼样品xrpd谱图
[0129]
图27是晶型b在水中溶解度测试中hplc图
[0130]
图28是晶型h的标准曲线
[0131]
图29是晶型h测试完水中溶解度后的滤饼样品xrpd谱图
[0132]
图30是晶型h在水中溶解度测试中hplc图
[0133]
图31是晶型b和晶型h在水中的溶解度曲线图
具体实施方式
[0134]
以下对本发明的具体实施方式进行详细的说明。应当理解的是,此处所描述的具体实施方式仅用于示例性地对本发明进行说明,并不用于限制本发明。
[0135]
实施例1化合物的制备
[0136]
1、制备i-1a-2
[0137][0138]
在干燥的圆底烧瓶中加入化合物1a(3.0g,17.8mmol),然后加入冰乙酸(40ml),0℃下依次缓慢滴加乙酸酐(6.8ml,71.4mmol,4.0eq)和浓硫酸(1.46ml,26.8mmol,1.5eq),移至室温反应过夜。加水,用乙酸乙酯萃取,水相用ea萃取一次,合并有机相,有机相用无水硫酸钠干燥,浓缩,真空抽干得到白色固体,直接用于下一步,产率80%。化合物1b 1
h nmr(400mhz,cdcl3)δ7.67(d,j=7.9hz,1h),7.30(t,j=8.0hz,1h),7.22(d,j=8.2hz,1h),3.89(s,3h),2.39(s,3h);m/z(es+)(m+na)
+
=233.1。
[0139]
在干燥的圆底烧瓶中加入化合物1b(3.01g,14.3mmol),然后加入乙醚(30ml),加入吡啶(1.16ml,14.3mmol,1.0eq)和二氯亚砜(1.25ml,17.2mmol,1.2eq),室温反应一小时。过滤,除去溶剂,剩余物直接用于下步反应。将所得酰氯溶于dmf(40ml),加入2-氨基-5-硝基噻唑(2.08g,14.3mmol,1.0eq)和三乙胺(12ml,85.9mmol,6.0eq),60℃下反应三个小时,低温下用1m hcl淬灭,乙酸乙酯萃取,有机溶液用1m hcl洗掉未完全反应的2-氨基-5-硝基噻唑,然后先后用饱和碳酸氢钠,水和饱和食盐水洗各一次,无水硫酸钠干燥,浓缩,用石油醚:乙酸乙酯=4:1重结晶得到黄色固体i-1a-2(123mg)。化合物i-1a-2 1
h nmr(400mhz,dmso)δ:13.60(br s,1h),8.71(s,1h),7.43-7.37(m,3h),3.84(s,3h),2.25(s,3h);m/z(es+)(m+na)
+
=360.1.2、采用制备化合物i-1a-2的方法制备得到下列化合物:
[0140]
化合物1:1h nmr(400mhz,dmso)δ13.60(br s,1h),8.70(s,1h),7.67(d,j=7.7hz,1h),7.59(d,j=7.6hz,1h),7.36(t,j=7.7hz,1h),2.28(s,3h),2.21(s,3h);m/z(es+)(m+na)
+
=344.0。
[0141]
化合物i-1a-3:1h nmr(400mhz,dmso)δ13.90(br s,1h),8.74(s,1h),8.37(dd,j=6.7,8.3hz,1h),8.21(dd,j=6.3,7.8hz,1h),7.73(t,j=8.0hz,1h),2.32(s,3h);m/z(es+)(m-h)-=351.0。
[0142]
化合物i-1a-4:1h nmr(400mhz,dmso)δ13.80(br s,1h),8.71(s,1h),8.00(d,j=8.0hz,1h),7.88(d,j=7.7hz,1h),7.42(t,j=8.0hz,1h),2.32(s,3h);m/z(es+)(m-h)-=
385.9。
[0143]
化合物i-1a-5:1h nmr(400mhz,dmso)δ13.78(br s,1h),8.73(s,1h),7.72-7.67(m,2h),7.54-7.48(m,1h),2.33(s,3h);
19
f nmr(400mhz,cdcl3)δ-128.53;m/z(es+)(m-h)-=324.0。
[0144]
化合物i-1a-6:1h nmr(400mhz,dmso)δ13.83(br s,1h),8.73(s,1h),7.91(dd,j=6.4,7.8hz,1h),7.85-7.82(m,1h),7.60(t,j=8.0hz,1h),2.32(s,3h);
19
f nmr(400mhz,cdcl3)δ-57.07;m/z(es+)(m-h)-=390.0。
[0145]
化合物i-1a-7:1h nmr(400mhz,dmso)δ13.90(br s,1h),8.74(s,1h),8.24-8.21(m,2h),7.69(t,j=7.8hz,1h),2.38(s,3h);m/z(es+)(m-h)-=331.0。
[0146]
化合物i-1a-8:1h nmr(400mhz,dmso)δ13.60(br s,1h),8.71(s,1h),7.69-7.66(m,2h),7.43(t,j=7.8hz,1h),2.28(s,3h),1.19(s,3h),1.17(s,3h);m/z(es+)(m-h)
+
=348.1。
[0147]
化合物i-1a-9:1h nmr(400mhz,dmso)δ13.53(br s,1h),8.70(s,1h),7.61(d,j=7.9hz,1h),7.26(d,j=8.0hz,1h),2.35(s,3h),2.29(s,3h),2.09(s,3h);m/z(es+)(m+na)
+
=358.0。
[0148]
化合物i-1a-10:1h nmr(400mhz,dmso)δ13.68(br s,1h),8.71(s,1h),7.90(d,j=2.2hz,1h),7.84(d,j=1.8hz,1h),2.28(s,3h),2.20(s,3h);m/z(es+)(m-h)-=399.9。
[0149]
化合物i-1a-11:1h nmr(400mhz,dmso)δ13.48(br s,1h),8.70(s,1h),7.69(d,j=8.8hz,1h),7.14(d,j=8.9hz,1h),3.93(s,3h),3.73(s,3h),2.30(s,3h);m/z(es+)(m+na)
+
=390.1。
[0150]
化合物i-1a-12:1h nmr(400mhz,dmso)δ13.62(br s,1h),8.71(s,1h),7.68(d,j=7.6hz,1h),7.61(d,j=7.6hz,1h),7.40(t,j=7.7hz,1h),2.58(dd,j=7.6,15.2hz,2h),2.28(s,3h),1.15(t,j=7.5hz,3h);m/z(es+)(m-h)-=334.1。
[0151]
化合物i-1b-12:将化合物i-1a-12(1.0g,2.98mmol)溶于thf(5ml)中,加入浓盐酸2ml,回流反应16h,冷却至室温,加入ea(100ml)萃取,有机相用饱和食盐水洗,无水硫酸钠干燥,过滤,浓缩,剩余物经硅胶柱层析纯化得i-1b-12(500mg,57%)。1h nmr(400mhz,dmso)δ8.75(s,1h),7.94(d,j=7.6hz,1h),7.41(d,j=7.0hz,1h),6.91(t,j=7.6hz,1h),2.64(q,j=7.4hz,2h),1.17(t,j=7.5hz,3h);m/z(esi)[m-h]-=292.1。
[0152]
实施例2抗乙肝病毒活性测试
[0153]
1、试验目的:本实验的目的是评价化合物的体外抗hbv活性。应用hepg2.2.15细胞评价受试化合物的体外抗乙肝病毒活性。通过实时荧光定量pcr(qpcr)方法检测hepg2.2.15细胞上清中的hbv dna的含量,elisa检测hbsag的含量测定化合物的抗hbv活性,同时celltiter-glo检测受试化合物对hepg2.2.15细胞活力影响。实验中采用lam作为参考化合物,监控实验质量。活性化合物再应用人原代肝细胞(phh)验证受试化合物体外抗hbv的药效。
[0154]
2、材料
[0155]
受试化合物和对照化合物:
[0156]
受试化合物由河南美泰宝生物制药有限公司提供,用100%dmso配制成20mm的母液。
[0157]
对照化合物:拉米夫定(lam),由药明康德提供,用100%dmso配制成20mm的母液。
[0158]
3、细胞株
[0159]
hepg2.2.15细胞由药明康德提供。
[0160]
4、试剂和耗材:
[0161]
本实验使用的主要试剂包括dna提取试剂盒(qiagen,货号51162),hbsag elisa试剂盒(货号:antu-cl 0310),faststart universal probe master(roche,货号04914058001),celltiter-glo检测试剂(promega,货号g7573),96孔板(costar 3599)和96well v型板(axygen wipp02280)。
[0162]
5、仪器:
[0163]
本实验使用的主要仪器包括荧光qpcr仪(applied biosystems,型号quantstudio(tm)6 flex system),酶标仪(biotek,型号synergy 2)。
[0164]
6、试验方法
[0165]
6.1细胞铺板和化合物处理
[0166]
第0天,将hepg2.2.15细胞以每孔60,000个细胞的密度接种到96孔板中,在37℃、5%co2培养过夜。第1天加入含不同浓度化合物的新鲜培养液,第4天起每天更换含不同浓度化合物的新鲜培养液,dmso终浓度为0.5%。抗hbv活性测试化合物最终测试浓度(μm)为:100、33.3、11.1、3.70、1.23、0.41、0.14。
[0167]
6.2、样品收集与检测
[0168]
抗病毒活性测试:第7天,收集细胞上清,应用dna提取试剂盒(qiagen-51162)提取上清中的dna,qpcr定量样品中的hbv dna。hbv质粒dna作为标准品,10倍系列稀释,标准品范围从1.0
×
107copies/μl至10copies/μl。pcr反应程序为:95℃10分钟,然后进入循环模式,95℃15秒,随后60℃,1分钟,共40个循环。依据标准曲线和各样品的ct值计算样品中的hbv dna含量。
[0169]
参照安图生物hbsag elisa kit说明书,应用酶标仪检测hbsag的含量。
[0170]
6.3数据处理及统计分析
[0171]
hbv dna抑制率(%)=(dmso对照组中的hbv dna含量-样品中的hbv dna含量)/dmso对照组中的hbv dna含量
×
100%
[0172]
hbsag抑制率(%)=(1-样品的hbsag值/dmso对照组hbsag值)
×
100%
[0173]
使用graphpad prism软件log(agonist)vs.response-variable slope(four parameters)分析方法处理数据得到ec
50
。
[0174]
6.4结果和结论
[0175]
受试化合物和对照化合物lam对细胞培养上清中hbsag、hbv dna的抑制结果见下表1:
[0176]
表1、化合物抗hbv活性(ec
50
)
[0177][0178]
对照化合物lam在hepg2.2.15细胞中抑制hbv dna的ec
50
值为0.11um,符合预期结果。受试化合物中,化合物i-1a-8、i-1a-9和i-1a-12三个化合物具有与化合物1相当的抑制hbv dna的活性,但是,它们抑制hbsag的活性是化合物1的2.5到9.5倍。因为现有临床核苷类乙肝药物,如恩替卡韦和富马酸丙酚替诺福韦(taf)都能够非常有效地抑制hbv dna,但是,还没有有效抑制hbsag的药物上市。所以,i-1a-8、i-1a-9和i-1a-12及其类似物的发现有十分重要意义。
[0179]
实施例3.抗新冠病毒活性测试
[0180]
(一)实验材料与试剂
[0181]
1.细胞系:vero-6细胞,本实验室保存;
[0182]
2.病毒株:2019-ncov(covid-19);
[0183]
3.测试药物,代表性化合物
[0184]
4.阳性对照药物:remdesivir,氯喹和lopinavir;
[0185]
5.试剂:dmem培养基(gibco),胎牛血清(gibco),双抗,胰酶,mtt(amresco)等;
[0186]
6.试剂盒:qiaamp viral rna mini kit(52906,qiagen),one step tb green primescript plus rt-pcr kit(perfect real time)(rr096a takara)
[0187]
7.耗材:细胞培养板,96孔酶标板等;
[0188]
8.仪器:多功能酶标仪,steponeplus荧光定量pcr仪,二氧化碳培养箱等。
[0189]
(二)实验步骤
[0190]
1.代表性化合物对细胞毒性测定
[0191]
利用mtt法检测待测化合物对vero-6细胞的毒性。mtt全称为3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide,是一种黄颜色的染料。mtt比色法,是一种检测细胞存活和生长的方法。其检测原理为活细胞线粒体中的琥珀酸脱氢酶能使外源性mtt还原为水不溶性的蓝紫色结晶甲瓒(formazan)并沉积在细胞中,而死细胞无此功能。10%sds(溶于0.01mol/l hcl溶液中)能溶解细胞中的甲瓒,用多功能酶标仪在570nm波长处测定其光吸收值,可间接反映活细胞数量。在一定细胞数范围内,mtt结晶形
成的量与细胞数成正比。通过检测不同干扰素浓度下570nm波长处光吸收值,即可计算该药物浓度下细胞的存活率,从而计算该药物的半数中毒浓度(cc
50
)。
[0192]
提前一天接种vero-6细胞于96孔板中,每孔1
×
104个(注意96孔板最边缘的孔勿用作实验孔,加pbs防止其他孔培养基挥发);观察细胞状态,达到约50%时,将待测化合物用含2%fbs的dmem培养基进行2倍比稀释后,以100μl/孔加至细胞板中,每种浓度设6个重复;同时设置对照组(不含药物组)和空白组(不含细胞组),置于37℃,5%co2的培养箱中培养;于加药后48h,每孔加入25μl mtt溶液(5mg/ml),继续培养4h后,每孔加入125μl 10%sds(溶于0.01mol/l hcl溶液中),轻轻吹打,放置2h,使结晶物充分溶解,以空白组调零,测定od570,按以下公式计算:存活率(%)=加药组od570/对照组od570
×
100%。同时计算药物的半数中毒浓度(cc50)。
[0193]
2.化合物对2019-ncov(covid-19)病毒的抑制效果评价
[0194]
在vero-6细胞模型上进行抗病毒活性测定,每次试验均设3复孔,共重复3次。
[0195]
1)于24孔细胞培养板每孔中接种5
×
104个vero-6细胞,在37℃,5%co2培养条件下,待汇合度达到60%时,按照感染复数moi为0.005,分别向每孔中加入200μl用含2%fbs的dmem培养基稀释后的2019-ncov(covid-19)病毒液,37℃、5%co2培养箱中吸附1h后,弃病毒液,将阳性对照药物和化合物用含2%fbs的dmem培养基从最大无毒浓度开始进行2倍比稀释,以500μl/孔加至细胞板中,同时设置对照组(不含药物组),于感染后48h收集上清病毒液。
[0196]
2)利用real-time rt-pcr(qrt-pcr)对收集的病毒进行rna定量:
[0197]
收集的上清病毒液分别取140μl,按照qiaamp viral rna mini kit试剂盒说明书进行rna提取。用one step tb green primescript plus rt-pcr kit(perfect real time)试剂盒进行qrt-pcr检测,
[0198]
反应体系总体积为20μl:10μl 2x one step tb green rt-pcr buffer4,1.2μl takara ex taq hs mix,0.4μl primescript plus rtase mix,rbd-qf1和rbd-qr1各0.8μl,0.4μl rox reference dye(50x),2μl病毒rna,4.4μl rnase free dh2o。反应参数为:反转录42℃5min,预变性95℃10s,pcr 40个循环包括变性95℃10s,退火和延伸60℃30s。
[0199]
3)计算每种浓度下的药物抑制率。抑制率(%)=1-实验组病毒rna拷贝数/不含药物组病毒rna拷贝数
×
100%。同时计算药物的半数有效浓度(ec
50
)和治疗指数(ti)=半数中毒浓度(cc
50
)/半数有效浓度(ec
50
)。
[0200]
(三)实验结果计算(表2)
[0201]
表2.代表性化合物抑制新冠病毒(covid-19)活性
[0202]
[0203][0204]
表2结果表明,本发明提供的一系列新化合物较硝唑尼特(1)的抗新冠活性显著提高。化合物i-1a-12和i-1b-12的抗新冠活性(ec
50
分别是1.48和1.30um)是化合物1(ec50是5.63)的3.80和4.33倍,是瑞德西韦的5.33和6.1倍。
[0205]
实施例4、动物药代动力学试验
[0206]
化合物以乙酸酯前药形式给药,在体内被酯酶水解释放出活性化合物,如化合物1转化为化合物2,以及化合物i-1a-12转化为化合物i-1b-12。
[0207][0208]
单次灌胃给予雄性sd大鼠受试化合物后,测定它们的代谢物在雄性sd大鼠体内的药代动力学性质。3只雄性sd大鼠单次灌胃给药。所有动物于给药后0.083h(5分钟)、0.167h(10分钟)、0.25h(15分钟)、0.5h(30分钟)、1h、2h、4h、8h和12h采集血浆样品。应用lc-ms/ms方法测定血浆样本中代谢产物(活性成分)的浓度。
[0209]
1、给药
[0210]
给药当天称量sd大鼠实际体重并计算给药体积。本试验中给药时雄性sd大鼠体重范围为230.65-259.01克。所有sd大鼠单次灌胃给药。
[0211]
2、样品收集与制备
[0212]
从试验动物颈静脉采集血液样本约0.2ml/动物/时间点,记录实际采血时间,给药后1小时以内的点在
±
1分钟范围内,其它时间在理论时间5%以内为可接受偏离范围。
[0213]
血样采集以后,立即转移至贴有标签的含k
2-edta(0.85-1.15mg)的商品化样品管中(江苏康健医疗用品有限公司),随后离心处理(3200g,4℃,10分钟)后取血浆。将血浆转移至预冷的离心管中,加入5倍体积含6种内标的acn沉淀,如100μl血浆样品+500μl的含6种内标的acn。沉淀后离心(4℃,12000rpm,10分钟)取上清(离心得血浆后20分钟内完成),在干冰中速冻,随后储存在-60℃或更低的超低温冰箱中,直到进行lc-ms/ms分析。
[0214]
3、分析方法
[0215]
化合物2和i-1b-12的血浆浓度使用lc-ms/ms方法进行测定。
[0216]
4、药代动力学数据分析
[0217]
使用winnonlin version 6.3(pharsight,mountain view,ca)药动学软件,以非房室模型对化合物2和i-1b-12的血浆药物浓度数据进行处理。使用线性对数梯形法计算相关药代动力学参数(表3)。
[0218]
表3、化合物1的药代动力学参数
[0219][0220][0221]
表3结果表明,雄性sd大鼠单次灌胃给予30mg/kg的化合物1后,化合物2的达峰浓度(c
max
)为19498
±
3831nmol/l,达峰时间(t
max
)出现在给药0.111
±
0.0485h。auc
0-last
为8778
±
4722nmol
·
h/l。
[0222]
表4、化合物i-1a-12的药代动力学参数
[0223][0224]
表4结果表明,雄性sd大鼠单次灌胃给予32.74mg/kg的i-1a-12后,化合物i-1b-12的达峰浓度(c
max
)为46329
±
5890nmol/l,达峰时间(t
max
)出现在给药后0.500h。auc
0-last
为109758
±
6007nmol
·
h/l。
[0225]
表3和表4的药代动力学参数比较可见,化合物2的半衰期(1.3h)和i-1b-12的(1.22h)相当;化合物i-1b-12的c
max
(46329)是化合物2的(19498)2.4倍;化合物i-1b-12的auc
0-last
(109758)是化合物2的(8778)的12.5倍。
[0226]
根据抑制乙肝表面抗原(hbsag)的活性和药代动力学的研究结果,上述实施例化合物不但抗病毒活性显著提高(提高2.5-10倍),而且有效药物的暴露量也有了巨大的提升(提升》10倍)。
[0227]
化合物i-1a-12的晶型研究
[0228]
晶型a
[0229]
将化合物i-1a-12固体(300mg)用异丙醇(3ml)和2-methf(1ml)重结晶,过滤,得到晶型a。tga、dsc和1h-nmr研究结果表明,晶型a为无水晶型,熔点为176.75℃。使用cu-kα辐射,晶型a在x射线粉末衍射图谱(图1)中2θ值=11.32
°
,12.01
°
,12.76
°
,16.39
°
,18.81
°
,19.61
°
,20.46
°
,22.74
°
,23.58
°
,24.07
°
,24.55
°
,24.97
°
,25.84
°
,27.15
°
,29.15
°
,29.86
°
,30.53
°
,35.24
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细峰数据见表5和图1。
[0230]
表5.晶型a的xrpd衍射峰数据
[0231]
pos.[
°
2θ]rel.int.[%]11.3226.9%12.0132.7%12.7625.4%16.3927.5%18.8165.3%19.6141.0%20.4637.4%22.7447.3%
23.58100.0%24.0716.9%24.5539.0%24.9713.1%25.8420.1%27.1553.0%29.1541.7%29.8615.6%30.5311.0%35.2425.0%
[0232]
晶型a在不同溶剂中,于25℃或30℃转化为晶型b。
[0233]
晶型b
[0234]
称取约300mg化合物i-1a-12固体于40ml玻璃瓶中,加入己烷(6.0ml)和etoac(4.0ml),在25℃条件下搅拌3天后过滤,将滤饼于50℃真空干燥12小时后,测定xrpd,所得晶体为晶型b。
[0235]
晶型a在室温/密封条件下放置20天后,转化成晶型b。tga、dsc和1h-nmr研究结果表明,晶型b为无水晶型。熔点为181.61℃。使用cu-kα辐射,晶型b的x射线粉末衍射图谱中2θ值=7.70
°
,9.32
°
,13.04
°
,15.41
°
,16.31
°
,17.48
°
,22.20
°
,23.25
°
,23.84
°
,24.29
°
,25.15
°
,25.57
°
,31.62
°
,39.20
°
,39.60
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细的峰数据见表6和图2。
[0236]
表6.晶型b的xrpd衍射峰数据
[0237]
pos.[
°
2θ]rel.int.[%]7.7054.0%9.3226.3%13.0451.8%15.4199.4%16.31100.0%17.4811.1%22.2016.8%23.2574.4%23.8416.5%24.2915.9%25.5726.8%31.6225.0%39.2027.0%39.6036.1%
[0238]
晶型c、d和e
[0239]
称取约40mg晶型a于样品瓶中,加入表7中所示溶剂,在50℃下使其溶解。随后将其过滤,将滤液降至约25℃,并在25℃下搅拌。对于未析出的样品继续搅拌1天。对仍未析出的
样品加入反溶剂。结果总结于表7。通过冷却结晶法,得到晶型b、晶型c、晶型d。晶型d在50℃真空干燥18小时转为晶型e。
[0240]
表7.冷却结晶实验参数
[0241][0242]
晶型c
[0243]
对晶型c进行tga、dsc、核磁等分析,其dsc结果有复杂的热现象。熔点峰所对应的tga温度有一个小的台阶失重(0.5%),且核磁结果显示样品有1.00%四氢呋喃残留。将晶型c通过dsc加热(10℃/min)到150℃,并在150℃下停留10min,将加热后的样品检测xrpd和1h nmr;结果表明晶型c在加热后,晶型未改变。晶型c的xrpd谱图与晶型b相似。form c为thf的溶剂化合物或含有thf溶剂残留的晶型b。使用cu-kα辐射,晶型c的xrpd图谱中2θ值=7.69
°
,9.32
°
,13.05
°
,15.43
°
,16.31
°
,17.46
°
,22.19
°
,22.52
°
,23.23
°
,23.84
°
,24.28
°
,24.86
°
,25.57
°
,31.62
°
,39.21
°
,39.61
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细的峰数据见表8和图3。
[0244]
表8.晶型c的xrpd衍射峰数据
[0245]
pos.[
°
2θ]rel.int.[%]7.6935.0%9.3219.7%13.0534.3%15.4372.3%16.31100.0%17.4612.1%22.1918.7%22.523.2%23.2333.7%23.8414.8%24.2817.0%24.8612.6%
25.5720.6%31.6224.2%39.2115.2%39.6130.0%
[0246]
晶型d
[0247]
对晶型d进行tga、dsc、核磁等分析,dsc结果显示复杂的热现象,第一个吸热峰所对应的温度范围内在tga曲线上有11.191%失重,且核磁结果显示样品有11.52%二氧六环残留,说明晶型d为溶剂化物,其在50℃下真空干燥18小时会转为晶型e或晶型g。使用cu-kα辐射,晶型d的x射线粉末衍射图谱中2θ值=9.59
°
,10.45
°
,10.68
°
,12.96
°
,15.30
°
,16.73
°
,17.25
°
,17.97
°
,18.26
°
,21.45
°
,23.42
°
,23.97
°
,23.42
°
,23.97
°
,24.45
°
,25.38
°
,25.69
°
,27.17
°
,27.84
°
,28.50
°
,29.26
°
,29.86
°
,30.20
°
,30.84
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细的峰数据见表9和图4。
[0248]
表9.晶型d的xrpd衍射峰数据
[0249]
pos.[
°
2θ]rel.int.[%]9.5918.2%10.4555.4%10.68100.0%12.9635.9%15.3017.2%16.7330.7%17.2512.3%17.9770.5%18.2635.2%21.4514.4%23.4216.7%23.9773.9%24.4533.9%25.3881.1%25.6910.2%27.1718.9%30.8416.4%
[0250]
晶型e
[0251]
对晶型e进行tga、dsc、核磁等分析,确认其为无水晶型。其onset为177.42℃,峰温度t
peak
为179.25℃。tga显示样品有0.4%的缓慢失重(为溶剂残留),核磁结果显示样品有0.43%的二氧六环溶剂残留。将晶型e通过dsc加热(10℃/min)到150℃,并在150℃下停留10min,将加热后的样品检测xrpd和1h nmr。结果表明晶型e在加热后转为晶型b;二氧六环溶剂极少量残留。使用cu-kα辐射,晶型e的xrpd谱中2θ值=9.63
°
,10.08
°
,12.40
°
,13.33
°
,14.10
°
,16.14
°
,23.38
°
,23.89
°
,24.30
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细的峰数据见表10和图5。
[0252]
表10.晶型e的xrpd衍射峰数据
[0253][0254][0255]
晶型f
[0256]
对晶型f进行tga、dsc、核磁等分析。其onset为178.59℃,t
peak
为179.46℃。dsc吸热峰温度对应的tga图谱有1.1%失重,且核磁结果有0.76%二氧六环残留。将晶型f通过dsc加热(10℃/min)到150℃,并在150℃下停留10min,将加热后的样品检测xrpd和1h nmr。结果表明晶型f在加热后,晶型未改变,二氧六环溶剂有残留。晶型f的xrpd谱图与晶型b相似,晶型f是溶剂化合物或含有溶剂残留的晶型b。使用cu-kα辐射,晶型f在x射线粉末衍射图谱中2θ值=7.64
°
,9.27
°
,12.42
°
,13.00
°
,15.38
°
,16.27
°
,17.42
°
,19.48
°
,22.14
°
,23.19
°
,23.79
°
,24.24
°
,24.81
°
,25.53
°
,31.56
°
39.17
°
,39.55
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细的峰数据见表11和图6。
[0257]
表11.晶型f的xrpd衍射峰数据
[0258]
pos.[
°
2θ]rel.int.[%]7.6444.4%9.2732.2%12.4211.4%13.0062.0%15.3877.0%16.27100.0%17.4218.5%19.4813.2%22.1424.5%23.1932.5%23.7914.3%24.2422.1%24.8116.7%25.5324.9%31.5620.2%39.1712.5%
39.5524.1%
[0259]
晶型h
[0260]
用晶型b制备晶型h:称取约20mg晶型b于8ml玻璃瓶中,加入乙醇(3.0ml)使其完全溶解,溶清后过滤,搅拌15min后析出固体。搅拌6hrs后过滤,将滤饼于50℃真空干燥14hrs;测定xrpd,所得晶体为晶型h。
[0261]
称取约800mg晶型b于250ml圆底烧瓶中,加入乙醇120.0ml在50℃条件下使其完全溶解后在空气中自然降温至25℃,析出固体较少,以3.33ml/min的速率加入溶剂正庚烷(60.0ml),搅拌1h后过滤,将滤饼于50℃真空干燥15hrs,检测xrpd证明所得晶体为晶型h。
[0262]
晶型d/e在正庚烷(25℃)中竞争打浆,或晶型g在乙醇中研磨3min,均得到晶型h。
[0263]
对晶型h进行tga、dsc、核磁等分析,确认其为无水晶型。其onset为179.01℃,t
peak
为181.10℃。tga与核磁结果显示样品无溶剂残留。xrpd使用cu-kα辐射,晶型h在x射线粉末衍射图谱中2θ值=9.74
°
,10.60
°
,14.04
°
,14.49
°
,15.7
°
,18.42
°
,19.70
°
,20.02
°
,25.73
°
,26.44
°
,29.33
°
的衍射角处有特征峰,其中2θ值误差范围为
±
0.2。更详细数据见表12和图7至图10。
[0264]
表12.晶型h的xrpd衍射峰数据
[0265]
pos.[
°
2θ]rel.int.[%]9.7412.2%10.6045.3%14.0457.7%14.4910.0%15.7529.6%18.4249.3%19.7023.4%20.0216.6%25.73100.0%26.4433.3%29.3314.3%
[0266]
晶型b和晶型h的稳定性研究
[0267]
称取适量的晶型b和晶型h于2ml小瓶中,在80℃下放置1天,在0天和1天进行取样分析其物理稳定性和化学稳定性。在25℃/60%rh和40℃/75%rh条件下放置7天,分别在0天,1天和7天取样分析其物理稳定性和化学稳定性。
[0268]
结果表明,晶型b和晶型h在80℃下放置一天和在25℃/60%rh、40℃/75%rh条件下放置7天后,物理化学性质稳定。结果见表13、表14和图11至图16。
[0269]
表13.晶型b和晶型h的稳定性研究
[0270][0271]
表14.晶型b和晶型h的80℃稳定性实验
[0272][0273]
晶型b和晶型h的研磨与压片实验
[0274]
为了验证晶型b和晶型h在研磨与压片条件下是否稳定,将晶型b和晶型h放入到玛瑙研钵中研磨3分钟,3分钟后分别检测其xrpd结果。如下:(1)干磨,称取约20mg,研磨3min,晶型未改变,结晶度略有下降;(2)湿磨,分别称取约20mg,分别加入20微升的水和乙醇,研磨3min,晶型未改变,结晶度略有下降。将晶型b和晶型h在20mpa下压片后,晶型未改变,结晶度略有下降。具体结果如图17和18所示。
[0275]
晶型b和晶型h的dvs测试
[0276]
对晶型b进行了dvs测试,结果显示样品在80%rh下增重为0.525%,因此晶型b略有引湿性;同时,对晶型b的dvs测试前后测定了xrpd,结果显示样品晶型在dvs测试前后是一致的。对晶型h进行了dvs测试,结果显示样品在80%rh下增重为0.007%,因此晶型h无引湿性;对晶型h的dvs测试前后测定了xrpd,结果显示样品晶型在dvs测试前后是一致的。采用另一批次晶型h样品对晶型h进行了dvs的复测,进一步验证了晶型h无引湿性。结果如图19至图24所示。
[0277]
晶型b在水中的溶解度测试
[0278]
标准溶液制备:称取10.12mg晶型b于20ml容量瓶中,加入稀释剂(水-乙腈(v/v)=1:1溶液),超声使固体溶解,再加入稀释剂定容至刻线。(溶液浓度为506μg/ml),标准曲线浓度见表15和图25。
[0279]
溶解度测试:称取约20mg的晶型b加入3ml水,配制2个平行样。分别在1小时,4小时,24小时取样,过滤。所得滤液进行hplc检测计算溶解度。结果如下表16、图26、图27和图31所示。
[0280]
表15.晶型b的标准曲线数据
[0281][0282]
表16.晶型b在水中的溶解度测试结果
[0283][0284]
晶型h在水中的溶解度测试
[0285]
称取约20mg的晶型h加入3ml水,配制2个平行样。分别在1小时,4小时,24小时取样,过滤。所得滤液利用hplc检测其溶解度,检测24小时后滤饼样品的xrpd。
[0286]
标准曲线浓度见表17和图28。
[0287]
溶解度测试结果如表18、图29至图31所示。
[0288]
表17晶型h的标准曲线数据
[0289][0290]
表18晶型h在水中的溶解度测试结果
[0291][0292]
八种晶型中晶型b和晶型h稳定性和引湿性性质相对较好,晶型b在水中的溶解度为1.31μg/ml,晶型h达到4.98-8.19μg/ml。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1