一种N-苯基烷氧基二苯并吖庚因类化合物、其制备方法及医药用途
一种n-苯基烷氧基二苯并吖庚因类化合物、其制备方法及医药用途
技术领域
1.本发明涉及药物化学和药物治疗学领域,具体涉及一类n-苯基烷氧基二苯并吖庚因类化合物。该类化合物可用于制备p-糖蛋白抑制剂、肿瘤多药耐药逆转剂。本发明还涉及该类化合物的制备方法以及含有它们的药物组合。
背景技术:
2.肿瘤细胞对多种结构和机制不同的抗癌药物的耐药性被称为多药耐药(multidrug resistance,mdr),它是肿瘤化疗的失败的主要原因(canc.lett.2014,347,159-166;mol.pharm.2011,8,1996-2011;methods mol.biol.2010,596,47-76.)。肿瘤mdr的产生机制非常复杂,包括细胞内酶异常改变(arch pharm res 2005,28,249-268.),细胞凋亡调控基因(尤其是p53和bcl-2)表达异常(cell,1993,74(6):957-967.),dna修复机制损伤(drug resist updat 2020,50,1-23.)等,其中研究最广泛的一个机制是atp结合式转运蛋白(atp-binding cassette(abc)transporters)的过度表达(nat.rev.cancer.2002,2,48-58;chem.rev.2009,109,2989-3011.);在abc转运蛋白家族成员中,p-糖蛋白(p-glycoprotein,p-gp)是第一个被发现与多药耐药相关的成员(discov.med.2006,6,18-23)。它能识别外源性抗癌药物并将它们排出癌细胞,导致细胞内药物积累减少。临床上许多化疗药物对p-gp介导的外排敏感,如紫杉醇、阿霉素、拓扑替康、达沙替尼、吉非替尼等(j.med.chem.2018,61,5108-5121)。研究表明,抑制p-gp可提高化疗药物在细胞内的积累,提高耐药细胞对化疗药物的敏感性,诱导肿瘤细胞凋亡,进而逆转mdr。因此,寻找和研究抑制p-gp的药物已成为克服mdr领域的研究热点之一。
3.目前已经开发了四代p-gp抑制剂(drug resist updat.2020,50,1-23)。然而,由于前三代p-gp抑制剂存在自身毒性高、靶点特异性差、药物与-药物相互作用或临床疗效不明显等原因,它们在临床研究中均以失败告终(j.med.chem.2020,63,5458-5476)。此外,p-gp在正常组织中同样表达,主要分布在肠上皮细胞、肾近曲小管细胞、肝胆管细胞膜,以及血脑、血睾和胎盘的上皮细胞(chemmedchem.2016,11,374-376),并通过外排泵参与分泌外源性物质和毒性代谢物的过程(expert opin.drug metab.toxicol.2008,4,205-223.)。当p-gp的正常功能被抑制后,抗癌药在体内的代谢和排泄受到影响,血药浓度水平提高,增加了其对正常组织的毒性。因此,迫切需要寻找选择性、高效、低细胞毒性的新型p-gp抑制剂来逆转肿瘤mdr。
4.从天然产物及衍生物中开发p-gp抑制剂已成为第四代抑制剂研发的新方向和重点。研究发现,许多天然产物如黄酮类、姜黄素类、五味子甲素及丙素等在抑制p-gp活性方面表现出的特有优势,如低毒、较好的耐受性等,使天然产物及其衍生物成为抑制剂开发新的希望和契机。
5.联苯双酯(bifendate)是一类源自五味子丙素的烷氧基联苯类化合物,是临床上用于治疗肝炎的降酶药物。研究表明,联苯双酯具有抗肿瘤活性,并且可以通过提高耐药肿
瘤细胞对p-gp底物的摄取量、减少底物外排,同时下调p-gp的表达,产生逆转肿瘤多药耐药的活性(invest.new drugs.2007,25,95-105.),并且bifendate和抗癌药联用,不影响抗癌药物的药代动力学性质,因此不会增加抗癌药的毒副作用。但是作为p-gp抑制剂,bifendate活性较低。
6.联苯双酯衍生物zg1142具有较好的p-gp抑制活性,且具有以下特点:1)该化合物自身毒性低,且不影响p-gp的基因及蛋白表达,提示其对细胞自身的重要生理机能损伤较小;2)该化合物在无毒剂量下(《ic
10
)能显著增加p-gp底物在高表达p-gp的耐药细胞k562/a02内的聚集,而不影响底物在敏感细胞k562内的聚集,提示可能是通过影响p-gp的功能来发挥逆转mdr活性;3)该化合物能够抑制p-gp atpase活性且其mdr逆转活性的持续时间长(》24h),提示该类肿瘤mdr逆转剂是非p-gp底物型抑制剂,具有第三代p-gp抑制剂的优点。值得注意的是,上述活性均显著强于先导化合物联苯双酯和经典的p-gp抑制剂维拉帕米(vrp)。
技术实现要素:
7.本发明的目的是提供一类n-苯基烷氧基二苯并吖庚因类化合物。药理实验证明,该类化合物具有良好的p-gp抑制活性,可以显著提高耐药肿瘤对化疗药的敏感性。
8.本发明的另一目的是提供一种n-苯基烷氧基二苯并吖庚因类化合物的制备方法。
9.本发明的第三个目的是提供一种n-苯基烷氧基二苯并吖庚因类化合物在制备与p-糖蛋白抑制功能药物、与逆转多药耐药性药物或抗肿瘤药物方面的应用。
10.本发明的目的可以通过以下措施达到:
11.通式i所示的n-苯基烷氧基二苯并吖庚因化合物或其药学上可接受的盐,
[0012][0013]
其中:
[0014]
x代表-co-、-nh-co-、-co-nh-、共价键、-nh-、s原子、-s(=o)
2-、-nh(so2)-或o原子;
[0015]
n代表0-6的整数;
[0016]
r代表羟基、羧基、硝基、氰基、卤素、c
1-c4烷基、c
1-c4烷氧基、苯基、取代苯基、苄基、取代苄基、环戊烷、环己烷、芳香杂环、取代芳香杂环、脂肪杂环或取代脂肪杂环;所述芳香杂环为喹啉、异喹啉、噻吩、呋喃、吡咯或吡啶;所述脂肪杂环为四氢喹啉、四氢异喹啉、六氢吡啶、四氢吡咯、哌嗪或吗啉;
[0017]
所述取代苯基、取代苄基、取代芳香杂环或取代脂肪杂环中的取代基选自下述基团中的一种或几种:c
1-c4烷基、c
1-c4烷氧基、羟基、硝基、氰基或卤素。
[0018]
在一种优选方案中,x代表-co-、-co-nh-、共价键或-nh-。
[0019]
在一种更优选方案中,x代表-co-、共价键或-nh-。
[0020]
在一种特别优选方案中,x代表共价键。
[0021]
在一种优选方案中,n代表1-4的整数。
[0022]
在一种更优选方案中,n代表1或2。
[0023]
在一种优选方案中,r代表羟基、羧基、溴、甲基、乙基、甲氧基、乙氧基、苯基、取代苯基、苄基、取代苄基、脂肪杂环或取代脂肪杂环。其中,所述脂肪杂环为四氢喹啉、四氢异喹啉、六氢吡啶、四氢吡咯、哌嗪或吗啉;所述取代苯基、取代苄基或取代脂肪杂环中的取代基选自下述基团中的一种或几种:甲基、乙基、甲氧基、乙氧基、氟、氯或溴。
[0024]
在一种更优选方案中,r代表羟基、羧基、溴、苯基、取代苯基、苄基、取代苄基、脂肪杂环或取代脂肪杂环;其中,所述脂肪杂环为四氢异喹啉、六氢吡啶、四氢吡咯、哌嗪或吗啉;所述取代苯基、取代苄基或取代脂肪杂环中的取代基选自下述基团中的一种或几种:甲氧基、乙氧基、氟、氯或溴。
[0025]
在一种特别优选方案中,r代表取代四氢异喹啉或六氢吡啶;所述取代四氢异喹啉中的取代基选自下述基团中的一种或几种:甲氧基或乙氧基。
[0026]
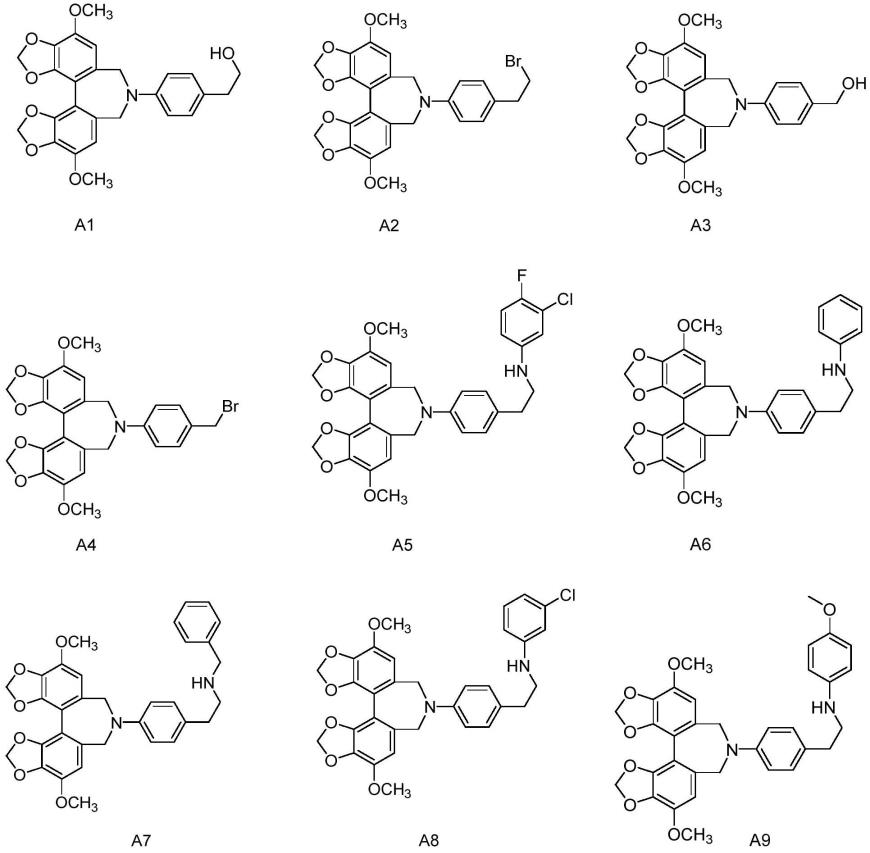
在一种更优选方案中,通式i所述化合物选自下列化合物:
[0027]
[0028]
[0029][0030]
在一种更优选方案中,通式i所述化合物选自下列化合物:
[0031][0032]
本发明公开了一种通式i所示化合物的制备方法,它包括如下步骤:
[0033]
a)当x代表共价键时,通式i所示化合物的制备方法包括如下步骤:
[0034]
将bifendate进行还原反应制备中间产物ii,中间产物ii与三溴化磷反应生成中间产物iii,中间产物iii与苯醇胺反应生成中间产物iv,中间产物iv与三溴化磷反应生成中间产物v,中间产物v与r-nh2反应生成通式i化合物;合成路线如下:
[0035][0036]
b)当x代表-co-时,通式i所示化合物的制备方法包括如下步骤:
[0037]
将bifendate进行还原反应制备中间产物ii,中间产物ii与三溴化磷反应生成中间产物iii,中间产物iii与对氨基苯乙酸乙酯反应生成中间产物vi,中间产物vi进行水解反应生成中间产物vii,中间产物vii与r反应生成通式i化合物;合成路线如下:
[0038][0039]
其中a)和b)的该方法中分别所涉及的各取代基的定义如上所述。
[0040]
本发明进一步公开了通式i所示化合物更加详细的制备方法,它包括如下步骤:
[0041]
a)当x代表共价键时,通式i所示化合物的制备方法包括如下步骤:
[0042]
将联苯双酯(bifendate)采用lialh4进行还原反应制备7,7'-二甲氧基-[4,4'-联苯并[d][1,3]二氧杂环戊烯-5,5'-二甲醇(中间产物ii),中间产物ii与三溴化磷在二氯甲烷中反应生成5,5'-双溴甲基-7,7'-二甲氧基-4,4'-联苯并[d][1,3]二恶唑(中间产物iii),中间产物iii与苯醇胺反应生成中间产物iv,中间产物iv与三溴化磷在吡啶存在下反应生成中间产物v,中间产物v与r-nh2反应生成通式i化合物;合成路线如下:
[0043][0044]
b)当x代表-co-时,通式i所示化合物的制备方法包括如下步骤:
[0045]
将联苯双酯(bifendate)采用lialh4进行还原反应制备7,7'-二甲氧基-[4,4'-联苯并[d][1,3]二氧杂环戊烯-5,5'-二甲醇(中间产物ii),中间产物ii与三溴化磷在二氯甲烷中反应生成5,5'-双溴甲基-7,7'-二甲氧基-4,4'-联苯并[d][1,3]二恶唑(中间产物iii),中间产物iii与对氨基苯乙酸甲酯在hatu、三乙胺存在下反应生成中间产物vi,中间产物vi在碱性条件下进行水解反应生成中间产物vii,中间产物vii与r在hatu、三乙胺存在下反应生成通式i化合物;合成路线如下:
[0046][0047][0048]
在上述制备方法中,由中间产物iii制备中间产物iv的过程中,采用的溶剂选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dma)、乙腈、四氢呋喃、二氯甲烷、1,2-二氯乙烷、氯仿中的一种或多种,优先选自乙腈;缚酸剂选自三乙胺、碳酸钾、氢氧化钠、碳酸钠、碳酸氢钠、乙酸钠或吡啶中的一种或多种,优先选自三乙胺。反应温度为0℃至回流温度。
[0049]
由中间产物v制备通式i化合物的过程中,采用的溶剂选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dma)、乙腈、四氢呋喃、吡啶、二氯甲烷、1,2-二氯乙烷、氯仿、甲苯或二氧六环中的一种或多种,优先选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺
(dma)、乙腈或四氢呋喃;反应温度为0℃至回流温度。
[0050]
由中间产物vii制备通式i化合物的过程中,采用的溶剂选自n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dma)、乙腈、四氢呋喃、二氯甲烷、1,2-二氯乙烷、氯仿中的一种或多种,优先选自n,n-二甲基甲酰胺(dmf);所采用的缩合剂为hatu、hbtu、hobt、edci或dcc,优先采用hatu;反应温度为室温。
[0051]
这些中间体或目标化合物均可按照常规分离技术加以纯化,并且根据需要将其转化为与可药用酸的加成盐。
[0052]
除非另外说明,在说明书和权利要求中使用的以下术语具有下面讨论的含义:
[0053]“药学上可接受的盐”表示保留母体化合物的生物有效性和性质的那些盐。这类盐包括:
[0054]
(1)与酸成盐,通过母体化合物的游离碱与无机酸或有机酸的反应而得,无机酸包括盐酸、氢溴酸、硝酸、磷酸、偏磷酸、硫酸、亚硫酸和高氯酸等,有机酸包括乙酸、三氟乙酸、丙酸、丙烯酸、己酸、环戊烷丙酸、羟乙酸、丙酮酸、草酸、(d)或(l)苹果酸、富马酸、马来酸、抗坏血酸、樟脑酸、苯甲酸、羟基苯甲酸、γ-羟基丁酸、甲氧基苯甲酸、邻苯二甲酸、甲磺酸、乙磺酸、萘-1-磺酸、萘-2-磺酸、对甲苯磺酸、水杨酸、酒石酸、柠檬酸、乳酸、肉桂酸、十二烷基硫酸、葡糖酸、谷氨酸、天冬氨酸、硬脂酸、扁桃酸、琥珀酸、戊二酸或丙二酸等。
[0055]
(2)存在于母体化合物中的酸性质子被金属离子代替或者与有机碱配位化合所生成的盐,金属例子例如碱金属离子、碱土金属离子或铝离子,有机碱例如乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、n-甲基葡糖胺、奎宁等。
[0056]“药物组合物”指将本发明中的化合物中的一个或多个或其药学上可接受的盐、溶剂化物、水合物或前药与别的化学成分,例如药学上可接受的载体,混合。药物组合物的目的是促进给药给动物的过程。
[0057]“药用载体”或“药学上可接受的载体”指的是对有机体不引起明显的刺激性和不干扰所给予化合物的生物活性和性质的药物组合物中的非活性成分,例如但不限于:碳酸钙、磷酸钙、各种糖(例如乳糖、甘露醇等)、淀粉、环糊精、硬脂酸镁、纤维素、碳酸镁、丙烯酸聚合物或甲基丙烯酸聚合物、凝胶、水、聚乙二醇、丙二醇、乙二醇、蓖麻油或氢化蓖麻油或多乙氧基氢化蓖麻油、芝麻油、玉米油、花生油等。
[0058]
“‑
c(o)
‑”
表示co基团或基团。
[0059]
“‑
nh-co
‑”
表示nhco基团或基团。
[0060]
“‑
co-nh
‑”
表示conh基团或基团。
[0061]
“‑
s(o)
‑”
表示so基团或基团。
[0062]“烷基”表示1-20个碳原子的饱和的脂烃基,包括直链和支链基团(本技术书中提到的数字范围,例如“1-20”,是指该基团,此时为烷基,可以含1个碳原子、2个碳原子、3个碳原子等,直至包括20个碳原子)。更优选的是,烷基是有1-10个碳原子的中等大小的烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基、叔丁基、戊基等。最好是,烷基为有1-8或1-6个
碳原子的低级烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基或叔丁基等。烷基可以是取代的或未取代的。当是取代的烷基时,该取代基优选是一或多个,更优选1-3个,最优选1或2个取代基。
[0063]“芳香杂环”或“杂芳基”表示5至12个环原子的单环或稠合环基团,含有一个、两个、三个或四个选自n、o或s的环杂原子,其余环原子是c,另外具有完全共轭的π电子系统。未取代的杂芳基地非限制性实例有吡咯、呋喃、噻吩、咪唑、噁唑、噻唑、吡唑、吡啶、嘧啶、喹啉、异喹啉、嘌呤、四唑、三嗪和咔唑。杂芳基可以是取代的或未取代的。当被取代时,取代基优选为一个或多个,更为优选为一个、两个或三个,进而更为优选一个或两个,独立地选自以下基团,包括:低级烷基、三卤烷基、卤素、羟基、低级烷氧基、巯基、(低级烷基)硫基、氰基、酰基、硫代酰基、o-氨基甲酰基、n-氨基甲酰基、o-硫代氨基甲酰基、n-硫代氨基甲酰基、c-酰氨基、n-酰氨基、硝基、n-磺酰氨基、s-磺酰氨基。优选的杂芳基可选地被一个或两个取代基取代,取代基独立地选自卤素、低级烷基、三卤烷基、羟基、巯基、氰基、n-酰氨基、单或二烷基胺基、羧基或n-磺酰氨基。
[0064]“羟基”表示-oh基团。
[0065]“烷氧基”表示-o-(未取代的烷基)和-o-(未取代的环烷基)。代表性实例包括但不限于甲氧基、乙氧基、丙氧基、丁氧基、环丙氧基、环丁氧基、环戊氧基、环己氧基等。
[0066]“卤素”表示氟、氯、溴或碘,优选为氟或氯。
[0067]
本发明的进一步提供了一种药物组合物,它以本发明所述的化合物或其药学上可接受的盐为活性成分或主要活性成分,辅以药学上可接受的载体。本发明化合物可以与其他抗肿瘤药物例如烷化剂(如环磷酰胺或顺铂)、抗代谢药(如5-氟尿嘧啶或羟基脲)、拓扑异构酶抑制剂(如喜树碱)、有丝分裂抑制剂(如紫杉醇或长春碱)、dna插入剂(如阿霉素)联合应用,另外还可以与放射治疗联合应用。通过和抗肿瘤药物联合治疗,增强耐药肿瘤细胞对抗癌药物的敏感性,从而有助于改善治疗效果。
[0068]
本发明的化合物或其药学上可接受的盐能够应用在制备与p-糖蛋白抑制功能药物、与逆转多药耐药性药物或抗肿瘤药物方面。
[0069]
采用本发明的技术方案,优势如下:
[0070]
本发明提供一类n-苯基烷氧基二苯并吖庚因化合物,该类化合物具有优良的p-糖蛋白抑制活性、肿瘤多药耐药逆转活性,临床上可作为肿瘤多药耐药逆转剂应用。此外,还开展了流式细胞实验及蛋白免疫印迹实验探究了活性化合物对p-gp外排功能及蛋白表达的影响,确定了活性化合物逆转肿瘤mdr的机制。
附图说明
[0071]
图1是rh123在k562/a02细胞中的蓄积情况;其中,药物的浓度分别为:a10 l:0.5μm;a10 m:2.0μm;a10 h:8.0μm;vrp:8.0μm;wk-x-34:8.0μm;实验平行三次,计算mean
±
sd,所有实验质与空白组对比具有显著性差异***p《0.001,a10高浓度h组和wk-x-34对比具有显著性差异###p《0.001;
[0072]
图2是p-gp在k562和k562/a02细胞中的表达;
[0073]
图3是p-gp在k562/a02细胞中与化合物作用后的表达;其中,以gapdh作为内参蛋白;药物浓度分别为:a10 l:0.5μm;a10 m:2.0μm;a10 h:8.0μm;vrp:8.0μm;wk-x-34:8.0μ
m;细胞在指定浓度的中处理48小时,收集上清液进行实验;
[0074]
图4是本发明代表化合物a10与p-gp晶体(pdb:4q9h)结构的分子对接图;其中,氨基酸残基用细棒表示、氢键和静电吸引力用虚线表示。
具体实施方式
[0075]
为了进一步阐明本发明,下面给出一系列实施例,这些实施例完全是例证性的,它们仅用来对本发明具体描述,不应当理解为对本发明的限制。
[0076][0077]
实施例1
[0078]
2-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯基)乙烷-1-醇(a1)
[0079]
步骤1:联苯双酯(5g,12mmol)溶于200ml四氢呋喃中,冰浴冷却至0℃,分批加入lialh4(1.357g,36mmol),自然升温至室温反应。tlc监测反应,反应结束后冰浴冷却至0℃,依次向反应液中加2ml水,6ml 15%naoh水溶液及2ml水,搅拌(灰白色混悬状)10min,加入适量的无水硫酸钠搅拌30-50min后抽滤,滤饼用甲醇多次冲洗。滤液减压浓缩后加水分散,乙酸乙酯萃取,有机层用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,减压浓缩,得白色固体7,7'-二甲氧基-[4,4'-联苯并[d][1,3]二氧杂环戊烯-5,5'-二甲醇(ii)(4.754g,产率95.08%)。该化合物为已知化合物。m.p.170-171℃(eur.j.med.chem.1992,27,353-358.172-173℃);esi-ms:m/z 385[m+na]
+
.
[0080]
步骤2:7,7'-二甲氧基-[4,4'-联苯并[d][1,3]二氧杂环戊烯-5,5'-二甲醇(ii)(500mg,1.38mmol)溶于4ml三氯甲烷,滴加三溴化磷(157μl,1.65mmol),反应液无色澄清透明,搅拌20min后,缓慢滴加2滴吡啶催化反应,室温条件下继续搅拌反应。tlc监测反应,2小时后反应结束后,加水分散,二氯甲烷萃取,有机层依次用蒸馏水、饱和碳酸氢钠溶液及饱和氯化钠溶液洗涤,无水硫酸钠干燥。过滤,浓缩,所得淡棕色油状物,静置后析出白色固体,加入无水乙醇抽滤,干燥后得到白色固体5,5'-双溴甲基-7,7'-二甲氧基-4,4'-联苯并[d][1,3]二恶唑(iii)(294mg,产率78.80%)。该化合物为已知化合物。m.p.143-145℃(yaoxue xuebao,1981,16(4),306-9.144-147℃);esi-ms:m/z 487.[m+h]
+
;
[0081]
步骤3:将5,5'-双溴甲基-7,7'-二甲氧基-4,4'-联苯并[d][1,3]二恶唑(iii)
(100mg,0.2mmol)用4ml乙腈完全溶解,加入苯乙醇胺(141mg,1.0mmol),五分钟后加入三乙胺(286μl,2.0mmol),反应液澄清透明。氮气保护下在金属浴中加热至40℃,冷凝回流。tlc监测反应,2小时后反应结束。减压浓缩除溶剂,得到无色油状液体,加水分散后乙酸乙酯萃取,有机层用饱和碳酸氢钠,蒸馏水和饱和氯化钠各洗涤一次,无水硫酸钠干燥后过滤,减压浓缩,得到类白色标题化合物a1(86.8mg,产率91.1%)。1h nmr(cdcl3,400mhz,δppm):7.20
–
7.07(m,2h),6.97
–
6.87(m,2h),6.53(s,2h),6.10(d,j=1.5hz,2h),6.00(d,j=1.5hz,2h),4.29(d,j=12.6hz,2h),3.89(s,6h),3.85
–
3.77(m,4h),2.80(t,j=6.5hz,2h).esi-ms:m/z 464.1[m+h]
+
;
[0082]
实施例2
[0083]
7-(4-(2-溴乙基)苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环己酮[4',5':5,6]苯并[1,2-e]氮杂卓(a2)
[0084]
步骤1-3:仿照实施例1所述步骤1-3的方法得到a1。
[0085]
步骤4:2-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯基)乙烷-1-醇(a1)(500mg,1.08mmol)用20ml二氯甲烷溶解,再加入三溴化磷(123.5μl,1.3mmol,密度:2.85g/ml),五分钟后加入0.2ml吡啶,氮气保护下在金属浴中逐渐升温至45℃,冷凝回流。2小时后tlc监测到原料反应完全,反应结束。加水分散,反应液用二氯甲烷萃取,有机层用饱和碳酸氢钠,蒸馏水各洗涤一次,无水硫酸钠干燥后过滤,减压浓缩,得到白色粉末状标题化合物(485.8mg,产率85.7%)。1h nmr(cdcl3,400mhz,δppm):7.13-7.03(m,2h),6.97
–
6.89(m,2h),6.76(s,2h),6.03(d,j=1.0hz,2h),5.98(d,j=1.0hz,2h),4.45(d,j=12.9hz,2h),3.79(s,6h),3.63(t,j=7.4hz,2h),3.53(d,j=12.8hz,2h),2.99(t,j=7.4hz,2h).esi-ms:m/z 526.0[m+h]
+
.
[0086]
实施例3
[0087]
4-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环丁烷[4',5':3,4]苯并[1,2-c][1,3]二氧杂环戊[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯甲醇(a3)
[0088]
仿照实施例1所述步骤1-3的类似方法,使用中间产物iii(500mg,1.0mmol),2-氨基苯甲醇(615.7mg,5.0mmol),三乙胺(1.43ml,10mmol)反应得到白色标题化合物(315mg,68.2%)。
[0089]1h nmr(400mhz,dmso-d6)δppm:7.16-7.07(m,2h),6.97
–
6.88(m,2h),6.74(s,2h),6.02(d,j=1.0hz,2h),5.96(d,j=1.0hz,2h),4.43(d,j=12.9hz,2h),4.34(d,j=5.5hz,2h),3.78(s,6h),3.52(d,j=12.8hz,2h);esi-ms:m/z 450.1[m+h]
+
.
[0090]
实施例4
[0091]
7-(4-溴甲基)苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环六醇[4',5':5,6]苯并[1,2-e]氮杂卓(a4)
[0092]
仿照实施例2所述步骤4的类似方法,使用化合物a3(300mg,0.64mmol),三溴化磷(73μl,0.77mmol,密度:2.85g/ml),0.2ml吡啶,反应得到白色标题化合物(236.5mg,69.3%)。
[0093]1h nmr(400mhz,cdcl3)δppm:7.22(d,j=8.2,2h),6.92(d,j=8.2hz,2h),6.52(s,2h),6.09(d,j=1.4hz,2h),5.99(d,j=1.4hz,2h),4.41
–
4.27(m,4h),3.87(s,6h),3.81
(d,j=12.6hz,2h).esi-ms:m/z 512.0[m+h]
+
.
[0094]
实施例5
[0095]
3-氯-n-(4-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧羰基[4',5':3,4]苯并[1,2-c][1,3]二氧杂环氧基[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯乙基)-4-氟苯胺(a5)
[0096]
步骤1-4:仿照实施例1所述步骤1-4的方法得到a2。
[0097]
步骤5:7-(4-(2-溴乙基)苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环己酮[4',5':5,6]苯并[1,2-e]氮杂卓(200mg,0.38mmol)溶解于15ml的n,n-二甲基甲酰胺溶液中,加入原料,3-氯-4-氟苯胺原料(110.6mg,0.76mmol),最后加入无水碳酸钾固体(262.6mg,1.9mmol),金属浴逐渐升温至90℃,开启搅拌,无水氯化钙保护,冷凝回流,9小时后tlc监测到原料反应完全,反应结束。加水分散,反应液用乙酸乙酯萃取,有机层用饱和氯化钠溶液,蒸馏水各洗涤一次,无水硫酸钠干燥后过滤,减压浓缩,得到白色粗品。经硅胶柱层析(二氯甲烷:甲醇=50:1,v/v)纯化后得白色粉末的标题化合物(76.8mg,产率34.2%)。
[0098]1h nmr(400mhz,dmso-d6)δppm:7.10-7.03(m,3h),6.91(d,j=8.5hz,2h),6.74(s,2h),6.64-6.59(m,1h),6.52-6.47(m,1h),6.02(d,j=1.0hz,2h),5.96(d,j=1.0hz,2h),5.83(s,1h),4.42(d,j=12.9hz,2h),3.78(s,6h),3.51(d,j=12.9hz,2h),3.12(s,2h),2.68(t,j=7.4hz,2h);esi-ms:m/z 591.1[m+h]
+
.
[0099]
实施例6
[0100]
n-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环胛[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯胺(a6)
[0101]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),苯胺(70.3μl,0.77mmol,密度:1.02g/ml),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(76.0mg,产率32.7%)。
[0102]1h nmr(400mhz,cdcl3)δppm:7.23-7.10(m,5h),6.97(s,2h),6.71(t,j=7.3hz,1h),6.62(d,j=8.0hz,2h),6.54(s,2h),6.11(d,j=1.4hz,2h),6.01(d,j=1.7hz,2h),4.42
–
4.21(m,4h),3.89(s,6h),3.37(t,j=7.0hz,2h),2.95
–
2.84(m,2h);esi-ms:m/z539.2[m+h]
+
.
[0103]
实施例7
[0104]
n-苄基-2-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环丁烷[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯基)乙烷-1-胺(a7)
[0105]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),苄胺(83.6μl,0.77mmol,密度:0.981g/ml),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(92.6mg,产率46.3%)。
[0106]1h nmr(400mhz,cdcl3)δppm:7.38-7.25(m,7h),7.12(d,j=8.2hz,2h),6.89(d,j=8.1hz,2h),6.52(s,2h),6.10(d,j=1.4hz,2h),5.99(d,j=0.8hz,2h),4.99(s,1h),4.37(d,j=6.0hz,2h),4.33
–
4.24(m,5h),3.88(s,7h),3.81(d,j=13.1hz,3h),2.87(t,j=7.2hz,2h);esi-ms:m/z 553.2[m+h]
+
.
[0107]
实施例8
[0108]
3-氯-n-(4-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环[4',5':3,4]苯并[1,
2-c][1,3]二氧杂环胛[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯乙基)苯胺(a8)
[0109]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),3-氯苯胺(80.5μl,0.77mmol,密度:1.22g/ml),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(73.4mg,产率33.7%)。
[0110]1h nmr(400mhz,cdcl3)δppm:7.14(d,j=8.0hz,2h),7.06(t,j=8.1hz,1h),6.94(s,2h),6.65(d,j=8.1hz,1h),6.58(d,j=2.3hz,1h),6.54(s,2h),6.46(d,j=8.5hz,1h),6.11(s,2h),6.01(s,2h),5.30(s,1h),4.36
–
4.22(m,4h),3.90(s,6h),3.35(t,j=7.2hz,2h),2.86(t,j=9.6hz,2h);esi-ms:m/z 573.1[m+h]
+
.
[0111]
实施例9
[0112]
n-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环戊[4',5':3,4]苯并[1,2-c][1,3]二氧杂环[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯乙基)-4-甲氧基苯胺(a9)
[0113]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),对甲氧基苯胺(94.8μl,0.77mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(86.6mg,产率40.0%)。
[0114]1h nmr(400mhz,cdcl3)δppm:7.11(d,j=8.5hz,2h),6.91(d,j=8.4hz,2h),6.83
–
6.73(m,2h),6.65
–
6.56(m,2h),6.53(s,2h),6.09(d,j=1.4hz,2h),5.99(d,j=1.5hz,2h),4.28(d,j=12.6hz,2h),3.88(s,6h),3.82(d,j=12.6hz,3h),3.74(s,3h),3.32(t,j=7.0hz,2h),2.84(t,j=7.0hz,2h);esi-ms:m/z 569.2[m+h]
+
.
[0115]
实施例10
[0116]
4-(4-(2-(6,7-二甲氧基-3,4-二氢异喹啉-2(1h)基)乙基)苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环[4',5':5,6]苯并[1,2-e]氮杂卓(a10)
[0117]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),6,7-二甲氧基-1,2,3,4-四氢异喹啉原料(146.7mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(116mg,产率47.7%)。
[0118]
m.p.234.0-236.5℃;1h nmr(400mhz,cdcl3)δppm:7.14-7.12(m,2h,2
×
ar-h),6.90
–
6.86(m,2h,2
×
ar-h),6.59(s,1h,ar-h),6.53(s,1h,ar-h),6.52(s,2h,2
×
ar-h),6.08(br,2h,-ch
2-),5.98(br,2h,-ch
2-),4.28(d,j=12.0hz,2h,-ch
2-),3.88(s,6h,2
×‑
och3),3.83(d,j=4.0hz,6h,2
×‑
och3),3.80(d,j=12.0hz,2h,-ch
2-),3.68(s,2h,-ch
2-),2.88
–
2.82(m,6h,3
×‑
ch
2-),2.78
–
2.74(m,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:148.08,147.72,147.40,145.86(2c),143.16(2c),134.98,129.97,129.71(2c),129.48(2c),126.30(2c),126.12(2c),115.48(2c),111.49,110.49,109.62,109.11(2c),101.84(2c),60.34,56.83,56.04(2c),56.00,55.65,52.28(2c),51.06,32.95,28.54.esi-hrms(tof):m/z[m+h]+calcd for c
37h38
n2o8,639.2701,found 639.2700;hplc purity 98.058%。
[0119]
实施例11
[0120]
4,10-二甲氧基-7-(4-(2-(6-甲氧基-3,4-二氢异喹啉-2(1h)基)苯基)-7,8-二氢-6h-[1,3]二氧噁并[4',5':3,4]苯并[1,2-c][1,3]二氧杂环戊[4',5':5,6]苯并[1,2-e]氮杂卓(a11)
[0121]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),6-甲氧基-1,2,3,4-四氢异喹啉原料(124.0mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到浅黄色粉末标题化合物(110mg,产率47.5%)。
[0122]
m.p.202.8-204.3℃;1h nmr(400mhz,cdcl3)δppm:7.15(d,j=8.0hz,2h,2
×
ar-h),6.96(d,j=8.0hz,2h,2
×
ar-h),6.90(d,j=8.0hz,2h,2
×
ar-h),6.71(d,j=8.0hz,2h,2
×
ar-h),6.65(s,1h,ar-h),6.53(s,2h,2
×
ar-h),6.10(s,2h,-ch
2-),5.99(s,2h,-ch
2-),4.29(d,j=12.0hz,2h,-ch
2-),3.89(s,6h,2
×‑
och3),3.81(d,j=12.0hz,2h,-ch
2-),3.67(s,3h,-och3),2.93
–
2.73(m,8h,4
×‑
ch
2-);
13
c nmr(101mhz,cdcl3)δppm:158.03,148.03,145.83(2c),143.16(2c),135.51(2c),134.95,130.20,129.72,129.49(2c),127.64,127.05(2c),115.49(2c),113.27(2c),112.15,110.47,109.03(2c),101.86(2c),60.62,56.79(2c),55.67,55.33,52.30,51.03(2c),33.07,29.48;esi-hrms(tof):m/z[m+h]
+
calcd for c
36h36
n2o7,609.2595,found 609.2595;hplc purity 95.661%。
[0123]
实施例12
[0124]
7-(2-(6,7-二甲氧基-3,4-二氢异喹啉-2(1h)基)乙基)苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环己烷[4',5':5,6]苯并[1,2-e]氮杂卓(a12)
[0125]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),6,7-二甲氧基-1,2,3,4-四氢异喹啉原料(146.7mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(121mg,产率49.7%)。
[0126]
m.p.179.3-182.3℃;1h nmr(400mhz,cdcl3)δppm:7.32
–
7.30(m,1h,ar-h),7.15
–
7.06(m,3h,3
×
ar-h),6.58(s,1h,ar-h),6.47(d,j=5.6hz,3h,3
×
ar-h),6.11(d,j=1.6hz,2h,-ch
2-),6.02(d,j=1.2hz,2h,-ch
2-),3.97(d,j=12.4hz,2h,-ch
2-),3.88(s,6h,2
×‑
ch3),3.84(s,3h,-ch3),3.82(s,3h,-ch3),3.71(s,2h,-ch
2-),3.62(d,j=12.4hz,2h,-ch
2-),3.13
–
3.10(m,2h,-ch
2-),2.86(s,6h,3
×‑
ch
2-);
13
c nmr(101mhz,cdcl3)δppm:151.64(2c),147.61,147.33,145.81(2c),143.25(2c),136.39,134.83(2c),130.46(2c),130.35,130.28,127.46,126.79,126.66,126.14,124.49,123.32,111.45,110.71,109.49,109.12(2c),101.84(2c),59.59,56.87(2c),55.97(2c),55.67,51.10,29.42,28.80;esi-hrms(tof):m/z[m+h]
+
calcd for c37h38n2o8,639.2701,found 639.2691;hplc purity 99.606%.
[0127]
实施例13
[0128]
4,10-二甲氧基-7-(4-(2-(哌啶-1-基)乙基)苯基)-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环戊[4',5':5,6]苯并[1,2-e]氮杂卓(a13)
[0129]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),六氢吡啶原料(64.7mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(98.4mg,产率64.8%)。
[0130]
m.p.245.5-247.0℃;1h nmr(400mhz,cdcl3)δppm:7.11(d,j=8.4hz,2h,2
×
ar-h),6.88(d,j=8.8hz,2h,2
×
ar-h),6.52(s,2h,2
×
ar-h),6.10(d,j=1.2hz,2h,-ch
2-),5.99(d,j=1.2hz,2h,-ch
2-),4.28(d,j=12.4hz,2h,-ch
2-),3.88(s,6h,2
×
ch3),3.80(d,j=12.8hz,2h,-ch
2-),2.78
–
2.74(m,4h,2
×‑
ch
2-),2.57
–
2.49(m,6h,3
×‑
ch
2-),1.65
–
1.62(m,4h,2
×‑
ch
2-),1.47(d,j=4.8hz,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:148.23,145.86(2c),143.17(2c),134.99(2c),129.66(2c),129.49(2c),128.69,115.48(2c),110.47(2c),109.09(2c),101.84(2c),60.68,56.83(2c),54.16(2c),52.21(2c),31.43,24.83(2c),23.70;esi-hrms(tof):m/z[m+h]+calcd for c
31h34
n2o6,531.2490,found 531.2535;hplc purity 98.044%.
[0131]
实施例14
[0132]
4,10-二甲氧基-7-(4-(2-(哌嗪-1-基)乙基)苯基)-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环戊[4',5':5,6]苯并[1,2-e]氮杂卓(a14)
[0133]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),哌嗪原料(65.4mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到白色标题化合物(78.8mg,产率38.99%)。
[0134]
m.p.205.3-206.2℃;1h nmr(400mhz,cdcl3)δppm:7.09-7.07(m,2h,2
×
ar-h),6.89
–
6.86(m,2h,2
×
ar-h),6.52(s,2h,2
×
ar-h),6.09(d,j=1.6hz,2h,-ch
2-),5.99(d,j=1.6hz,2h,-ch
2-),4.28(d,j=12.8hz,2h,-ch
2-),3.88(s,6h,2
×‑
ch3),3.80(d,j=12.8hz,2h,-ch
2-),3.14(t,j=4.8,hz),2.74
–
2.70(m,6h,3
×‑
ch
2-),2.66
–
2.62(m,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:148.11,145.86(2c),143.15(2c),134.98(2c),129.67(2c),129.42(2c),129.36,115.42(2c),110.47(2c),109.09(2c),101.85(2c),60.43,56.83(2c),52.22(2c),51.32(2c),44.44(2c),32.38;esi-hrms(tof):m/z[m+h]+calcd for c
30h33
n3o6,532.2442,found 532.2391;hplc purity 95.020%.
[0135]
实施例15
[0136]
4,10-二甲氧基-7-(4-(2-(吡咯烷-1-基)乙基)苯基)-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环戊醇[4',5':5,6]苯并[1,2-e]氮杂卓(a15)
[0137]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),四氢吡咯原料(54.7mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到浅黄色粉末状的标题化合物(85.3mg,产率43.4%)。
[0138]
m.p.310.9-312.0℃;1h nmr(400mhz,cdcl3)δppm:7.12(d,j=8.0hz,2h,2
×
ar-h),6.87(d,j=8.8hz,2h,2
×
ar-h),6.50(s,2h,2
×
ar-h),6.07(d,j=1.6hz,2h,-ch
2-),5.97(d,j=1.6hz,2h,-ch
2-),4.85(s,2h,-ch
2-),4.27(d,j=12.8hz,2h,-ch
2-),3.87(s,6h,2
×
ch3),3.77(d,j=12.4hz,2h,-ch
2-),3.46
–
3.13(m,6h,3
×‑
ch
2-),2.13(s,4h,2
×‑
ch
2-);
13
c nmr(101mhz,cdcl3)δppm:148.67,145.92(2c),143.16(2c),135.06(2c),129.55(2c),129.48(2c),125.50,115.44(2c),110.44(2c),109.12(2c),101.87(2c),57.21,56.88(2c),53.92(2c),52.01(2c),31.22,23.50(2c);esi-hrms(tof):m/z[m+h]+calcd for c30h32n2o6,517.2333,found 517.2306;hplc purity 97.027%。
[0139]
实施例16
[0140]
4,10-二甲氧基-7-(4-(2-吗啉乙基)苯基)-7,8-二氢-6h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环胛[4',5':5,6]苯并[1,2-e]氮杂卓(a16)
[0141]
仿照实施例5所述步骤4的类似方法,使用化合物a2(200mg,0.38mmol),吗啉原料(66.2mg,0.76mmol),无水碳酸钾固体(262.6mg,1.9mmol),反应得到浅黄色粉末状的标题化合物(96.5mg,产率47.7%)。
[0142]
m.p.217.9-219.5℃;1h nmr(400mhz,cdcl3)δppm:7.10(d,j=12.0hz,2h,2
×
ar-h),6.88(d,j=12.0hz,2h,2
×
ar-h),6.52(s,2h,2
×
ar-h),6.08(d,j=1.2hz,2h,-ch2-),5.98(d,j=2.4hz,2h,-ch
2-),4.28(d,j=12.8hz,2h,-ch
2-),3.88(s,6h,2
×‑
ch3),3.8(d,j=12.4hz,2h,-ch
2-),3.74(t,j=4.8hz,2h,-ch
2-),2.76
–
2.72(m,2h,-ch
2-),2.60
–
2.51(m,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:148.04,145.86,143.15(2c),134.97(2c),129.84(2c),129.70(2c),129.44(2c),115.43(2c),110.48(2c),109.09(2c),101.84(2c),67.04(2c),61.19,56.80(2c),53.81(2c),52.24(2c),32.35;esi-hrms(tof):m/z[m+h]
+
calcd for c
30h32
n2o7,533.2282,found 533.2231;hplc purity 95.070%。
[0143]
实施例17
[0144]
4-(6,7-二甲氧基-3,4-二氢异喹啉-2(1h)基)甲基苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环丁烷[4',5':5,6]苯并[1,2-e]氮杂卓(a17)
[0145]
仿照实施例5所述步骤4的类似方法,使用化合物a4(200mg,0.39mmol),6,7-二甲氧基-1,2,3,4-四氢异喹啉原料(150.7mg,0.78mmol),无水碳酸钾固体(276.4mg,2.0mmol),反应得到浅黄色粉末的标题化合物(110.1mg,产率45.1%)。
[0146]
m.p.262.4-264.0℃;1h nmr(400mhz,cdcl3)δppm:7.27(d,j=8.4hz,2h,2
×
ar-h),6.91(d,j=8.8hz,2h,2
×
ar-h),6.58
–
6.48(m,4h,4
×
ar-h),6.09(d,j=1.6hz,2h,-ch
2-),5.99(d,j=1.2hz,2h,-ch
2-),4.33(d,j=12.4hz,2h,-ch
2-),3.88(s,6h,2
×‑
ch3),3.81(br,8h,2
×‑
ch3,-ch
2-),3.60(d,j=7.6hz,2h,-ch
2-),3.53(s,2h,-ch
2-),2.81(d,j=5.2hz,2h,-ch
2-),2.73(d,j=4.4hz,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:148.78,147.62,147.30,145.89,143.17(2c),135.01(2c),130.32(2c),129.71(2c),127.68,126.77,126.32,114.91(2c),111.55,110.50(2c),109.67,109.10(2c),101.86(2c),62.23,56.82(2c),56.00(2c),55.59,53.53,52.10(2c),50.71,28.69;esi-hrms(tof):m/z[m+na]
+
calcd for c
36h36
n2o8,647.2364,found 647.2291;hplc purity 96.514%.
[0147]
实施例18
[0148]
4,10-二甲氧基-7-(4-(哌啶-1-基甲基)苯基)-7,8-二氢-6h-[1,3]二氧杂环噁[4',5':3,4]苯并[1,2-c][1,3]二氧杂环己烷[4',5':5,6]苯并[1,2-e]氮杂卓(a18)
[0149]
仿照实施例5所述步骤4的类似方法,使用化合物a4(200mg,0.39mmol),六氢吡啶原料(66.4mg,0.78mmol),无水碳酸钾固体(276.4mg,2.0mmol),反应得到浅黄色粉末的标题化合物(85.2mg,产率42.2%)。
[0150]
m.p.196.7-198.7℃;1h nmr(400mhz,cdcl3)δppm:7.30
–
7.29(m,2h,2
×
ar-h),6.90
–
6.89(m,2h,2
×
ar-h),6.52(s,2h,2
×
ar-h),6.08(d,j=1.6hz,2h,-ch
2-),5.98(d,j=1.2hz,2h,-ch
2-),4.32(d,j=12.8hz,2h,-ch
2-),3.87(s,6h,2
×‑
ch3),3.80(d,j=10.4hz,2h,-ch
2-),3.71(d,j=12.8hz,2h,-ch
2-),2.67(s,4h,2
×‑
ch
2-),1.78(t,j=6.0hz,4h,2
×‑
ch
2-),1.50(s,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:149.47,145.90(2c),143.18(2c),135.05(2c),131.65(2c),129.46(2c),121.87,114.66(2c),110.40(2c),109.04(2c),101.90(2c),62.04,56.85(2c),53.34(2c),51.78(2c),24.23(2c),23.28;esi-hrms(tof):m/z[m+na]
+
calcd for c
30h32
n2o6,539.2153,found 539.2108;hplc purity 95.615%.
[0151]
实施例19
[0152]
7-(6,7-二甲氧基-3,4-二氢异喹啉-2(1h)基)甲基苯基)-4,10-二甲氧基-7,8-二氢-6h-[1,3]二氧杂环[4',5':3,4]苯并[1,2-c][1,3]二氧杂环六[4',5':5,6]苯并[1,2-e]氮杂卓(a19)
[0153]
仿照实施例5所述步骤4的类似方法,使用间位苄溴代原料(200mg,0.39mmol),6,7-二甲氧基-1,2,3,4-四氢异喹啉(66.4mg,0.78mmol),无水碳酸钾固体(276.4mg,2.0mmol),反应得到白色标题化合物(85.2mg,产率42.2%)。
[0154]1h nmr(400mhz,cdcl3)δppm:7.25
–
7.21(m,1h,ar-h),7.04(t,j=2.8hz,1h,ar-h),6.89
–
6.86(m,1h,ar-h),6.83
–
6.81(m,1h,ar-h),6.60
–
6.50(m.4h,4
×
ar-h),6.09(t,j=1.2hz,2h,-ch
2-),5.99(t,j=1.2hz,2h,-ch
2-),4.37(d,j=12.8hz,-ch
2-),3.87(s,6h,2
×‑
ch3),3.84
–
3.80(m,8h,2
×‑
ch3,-ch
2-),3.70
–
3.58(m,4h,2
×‑
ch
2-),2.83(t,j=6.0hz,2h,-ch
2-),2.75(t,j=5.6hz,2h,-ch
2-);
13
c nmr(101mhz,cdcl3)δppm:153.01,149.67,147.59,147.28,145.84(2c),143.15(2c),134.96(2c),129.72(2c),129.11,126.17,119.35,115.70,113.97,111.41,110.48,109.53,109.00(2c),101.89(2c),63.27,56.77(2c),55.99(2c),55.85,55.82,51.99(2c),50.80,29.81,28.76;esi-hrms(tof):m/z[m+h]
+
calcd for c
36h36
n2o8,625.2544,found 625.2587;hplc:purity 97.18%。
[0155][0156]
实施例20
[0157]
2-(4-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环戊[4',5':3,4]苯并[1,2-c][1,3]二氧杂环胛[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯基)乙酸乙酯(b1)
[0158]
步骤6:将5,5'-双溴甲基-7,7'-二甲氧基-4,4'-联苯并[d][1,3]二恶唑(ⅲ)(300mg,0.62mmol)用12ml乙腈完全溶解,加入4-氨基苯乙酸乙酯(333.3mg,1.86mmol),五分钟后加入三乙胺(517μl,3.72mmol),反应液澄清透明。氮气保护下在金属浴中加热至40℃,冷凝回流。tlc监测反应,5小时后反应结束。减压浓缩除溶剂,得到无色油状液体,加水分散后乙酸乙酯萃取,有机层用饱和碳酸氢钠,蒸馏水和饱和氯化钠各洗涤一次,无水硫酸钠干燥后过滤,减压浓缩,得到类白色标题化合物(269.1mg,产率86.3%)。
[0159]1h nmr(400mhz,cdcl3)δppm:7.20(d,j=8.0hz,2h),7.07(d,j=8.0hz,1h),6.94(s,1h),6.66
–
6.63(m,1h),6.54(s,1h),6.11(d,j=4.5hz,2h),6.01(d,j=8.0hz,2h),4.44-4.25(m,2h),4.24-3.96(m,4h),3.89(s,6h),3.52(d,j=21.5hz,2h),1.26(t,3h);esi-ms:m/z 506.5[m+h]
+
.
[0160]
实施例21
[0161]
2-(4,10-二甲氧基-6,8-二氢-7h-[1,3]二氧杂环胛[4',5':3,4]苯并[1,2-c][1,3]二氧杂环[4',5':5,6]苯并[1,2-e]氮杂卓-7-基)苯乙酸(b2)
[0162]
步骤7:取b1(250mg,0.5mmol)化合物于100ml茄形瓶中,用40ml乙醇溶解后。加入
8溶液,放入振荡器上轻轻混匀,再放入37℃培育箱培育3h,用酶标仪检测450nm处od值,计算细胞相对成活率,实验需重复3次,取平均值。根据od值制作细胞增殖曲线,评价细胞增殖能力的变化。实验结果如表1所示。
[0171]
抑制率(%)=(阴性对照组od值-实验组od值)/阴性对照组od值
×
100%;
[0172]
根据抑制率,采用logit法计算半数抑制浓度ic
50
值。实验重复3次,数据表示为mean
±
sd。
[0173]
表1本发明代表化合物对耐药肿瘤细胞k562/a02增殖的抑制活性
[0174][0175][0176]
结果发现抗肿瘤药adr对k562细胞抑制活性较强(ic
50
=0.001
±
0.01μm),对k562/a02细胞抑制活性显著降低,减弱了约3000倍(ic
50
=3.44
±
0.61μm),表明k562/a02细胞对adr产生了耐药。部分化合物在体外显示一定的自身细胞毒性(ic
50
《10μm),其中连接臂含有苯环结构的部分目标化合物a10、a11、a12、a15、a17和a19的安全性最高(ic
50
》95μm)。基于此,我们选择无毒浓度开展上述化合物的逆转肿瘤mdr活性评价实验。
[0177]
2、化合物的肿瘤mdr逆转实验
[0178]
adr单独或者和无毒浓度的化合物一起与k562/a02细胞共育采用cck-8法,对目标化合物进行逆转实验。选择阿霉素(adr)、zg1142(6-[2-(3,4-亚甲二氧基)苯基]乙基-3,9-二甲氧基-1,2-亚甲二氧基-10,11-亚甲二氧基-6,7-二氢-5h-二苯并[c,e]吖庚因)和第三代p-gp抑制剂wk-x-34为阳性对照药,实验结果如表2所示。
[0179]
表2本发明代表化合物在2.0μmol
·
l-1
浓度下对阿霉素的逆转活性
[0180]
[0181]
注:a.rf:药物敏感性的逆转倍数,rf=阿霉素的ic
50
/与目标化合物联合使用后的阿霉素的ic
50
值。
[0182]
实验结果表明,化合物a10(rf=93.17)、a17(rf=374.09)、a18(rf=113.14)和a19(rf=92.10)可以很大程度恢复k562/a02细胞对adr的敏感性,且活性优于先导物zg1142(rf=20.58)。由此可知,化合物a10、a17、a18和a19能够增加k562/a02细胞对adr的敏感程度。其中,化合物a17的作用最为显著,明显高于阳性对wk-x-34(rf=81.90)和zg1142(rf=20.58)。再结合细胞毒性实验结果,综合考虑化合物自身毒性的影响,可选择安全性高的化合物a10用于后续的机制实验研究。
[0183]
3、量效关系实验
[0184]
采用cck-8法测定化合物逆转k562/a02细胞阿霉素耐药的ec
50
值,评定目标化合物逆转阿霉素耐药的逆转活性和量效关系,测定了k562/a02细胞的ec
50
值,以确定有效浓度,实验结果如表3所示。
[0185]
表3化合物a10在梯度浓度下对k562/a02细胞的抗肿瘤mdr活性
[0186][0187]
注:a.rf:药物敏感性的逆转倍数,rf=阿霉素的ic
50
/与目标化合物联合使用后的阿霉素的ic
50
值。
[0188]
如表3所示,zg1142相同浓度下逆转活性均小于化合物a10,在0.5μm时两者均表现出较弱的调节活性,而化合物a10的逆转活性为zg1142的两倍。a10在浓度降至0.25μm时仍
表现较强的逆转多药耐药活性(rf=4.79),并具有明显的剂量依赖活性。此外,由graphpad prism5.0软件根据剂量-效应曲线计算得出化合物a10的ec
50
值为0.7443nm。结果表明,化合物a10具有明显的增强p-gp过表达细胞的对抗癌药物底物敏感性的潜力,且呈剂量依赖关系。
[0189]
4、活性化合物a10对rh123蓄积和外排的影响
[0190]
抑制p-gp的转运功能或者降低p-gp表达均可以逆转p-gp介导的肿瘤mdr。为了评价探究活性化合物逆转肿瘤mdr的作用机制,我们首先采用流式细胞分析法,检测活性化合物对罗丹明123(rh123,p-gp的荧光底物)在耐药k562/a02细胞内积累的影响,初步评价其对p-gp外排功能的作用。选择经典抑制剂vrp和wk-x-34为阳性对照。
[0191]
实验结果表明(图1),化合物a10能剂量依赖性地增加rh123在k562/a02细胞内的水平。值得注意的是,a10在中等浓度下超过高浓度的vrp,高浓度下低于wk-x-34。上述结构表明,化合物a10可以影响p-gp的功能。
[0192]
5、化合物a10对p-gp蛋白及基因表达的影响
[0193]
接着,采用western blotting法检测活性化合物a10对p-gp蛋白表达的影响。选择vrp、wk-x-34作为阳性对照。实验结果表明p-gp在敏感k562细胞中p-gp表达含量较少,而耐药k562/a02细胞中高表达p-gp(图2)。化合物a10与k562/a02共育后不影响p-gp蛋白的表达(图3)。上述实验结果初步表明,化合物a10是通过影响p-gp外排泵的功能,进而逆转p-gp介导的肿瘤mdr。
[0194]
6、化合物分子对接分析
[0195]
将活性最好的化合物与人类同系物p-gp的药物结合口袋(pbd id:4q9h)的进行对接,通过分析存在的相互作用来解释活性结果,结果见图4。
[0196]
图4表明化合物a10结构上的四氢异喹啉和烷氧基二苯并吖庚因母核上取代的甲氧基中的氧原子分别与thr-199、ser-196、phe-983形成氢键相互作用,结合质子四氢异喹啉部分与glu-875形成静电吸引力;作为连接臂的苯环与met-986形成π-硫相互作用;n-苯基烷氧基二苯并吖庚因母核与周围苯丙氨酸和酪氨酸形成的疏水空腔的亲和能力合较好。
[0197]
以上药理学数据显示,本发明化合物具有优异的p-gp抑制作用,能显著逆转p-gp介导的mdr。其中,化合物a10、a17、a18和a19对p-gp的抑制作用显著强于阳性对照药第三代p-gp抑制剂wk-x-34及我们课题组前期发现的活性最好的烷氧基联苯类化合物zg1142。并且对化合物a10进行了部分机制研究,结果表明化合物a10是一个自身毒性低、逆转肿瘤mdr活性高、不影响p-gp表达、抑制p-gp外排功能的潜在的p-gp抑制剂。
[0198]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可能对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1