一种聚醚酮酮树脂的制备方法与流程
1.本发明属于高分子材料合成技术领域,涉及一种聚醚酮酮树脂的制备方法。
背景技术:
2.聚芳醚酮(paek)特种工程塑料,具有卓越的机械物理性能,而在聚芳醚酮聚合物中,与其他类高性能塑料相比,聚醚酮酮(pekk)是目前特种工程塑料中耐热性能最高,综合性能最好的树脂材料,长期使用耐热性超过250℃。此外,pekk在高温下还具有优良的力学性能、电绝缘性、耐化学品性和耐辐射等特点。pekk优异的综合性能使其广泛用于航空航天、电子信息、机械医疗设备等领域。pekk可以是无定形的或半结晶的,两种类型通常都具有较高的玻璃化转变温度(tg),而半结晶形还具有较高的熔融温度(tm)。聚(醚酮酮)系列还因具有优异的机械性能、化学惰性和抗应力开裂性而特别受到关注,可用于航空航天领域和许多其他广泛的工业应用中,包括制备热塑性复合材料,同时,还可用于制备生物医学植入物材料。
3.通用术语通过参考重复单元的结构(在高分子化学中是标准的)来命名此类聚合物,并根据醚(以“e”表示)和酮(以“k”表示)的序列命名。在重复单元中,例如,基本上将由重复单元:-ar-o-ar-(c=o)-ar-(c=o)-构成的聚合物称为“pekk”。可以使用friedel-crafts亲电聚合反应来制备聚醚酮酮pekk。
[0004][0005]
通常,在聚醚酮中连接醚和酮键的桥接是芳香苯环单元,其本身被1,4或1,3-二取代(1,2-二取代是有可能的,但不常见)。所有1,4-取代的聚醚酮酮均具有较高的结晶度。另外,随着酮与醚的比率增加,聚合物的熔融温度tm增加,1,4-pekk的tm约为395℃,显示出35%至40%之间的极限结晶度。尽管可以通过任何一种亲电方法轻松合成1,4-pekk,但高熔融温度的1,4-pekk由于难以在溶液中保留足够长的时间以形成高分子量产物,因此通过亲电方法合成高分子量的1,4-pekk变得困难。众所周知,可以通过在结构中加入1,3单元来控制所得聚醚酮的熔融温度tm,从而使1,4和1,3单元的合适混合物可以生成无定形或结晶度低产品。虽然所有的1,4-pekk都难以熔融加工,但也可以从浓硫酸,二氯乙酸等溶剂中进行溶液加工,但这需要高昂的成本,显然这在商业化、工业化生产中是不可取的,因为它需要定制特种专用设备来防止酸性溶液的侵蚀,同时也对环境有更高的要求来满足安全生产。
[0006]
对于体内使用pekk材料必须进一步满足生物相容性的要求,理想状态下,合成的聚合物中应无未反应的单体、催化剂残留物或其他反应组分,即需要聚合物的高纯度来满
足生物相容性的要求。事实上,聚合物中夹杂着杂质,这些杂质还会导致加工过程中聚合物的熔体不稳定。在进一步加工成型期间,这可能是一个严重的问题,因为不稳定性会导致制造过程中的性能变化,从而可能影响制品使用中的性能。对于结构和性能要求较高的应用,例如航空航天等,是非常不希望的。
[0007]
芳香族聚醚酮通常通过亲核或亲电聚合反应制备。例如,由英国victrex提供的聚合物peek被认为是通过高温亲核过程合成的,工艺为将4,4-二氟二苯甲酮、对苯二酚、碳酸盐在二苯砜熔融环境下,在280~340℃的温度下进行缩聚反应,如下所示:
[0008][0009]
其亲核反应是溶液缩聚反应,其中通过聚合物保留在溶液中(例如,在高温下的二苯砜中),使增长的聚合物链保持在反应状态。相反,亲电反应不是真正的溶液缩聚反应,因为随着反应链的增长,反应生成的聚合物从溶液中析出来。与正常的沉淀不同,颗粒会聚集形成难以处理的团块。
[0010]
cn109608627中公开了一种聚醚酮酮的生产工艺,其引入了路易斯碱,并通过低温的亲电反应的方法进行合成聚醚酮酮,众所周知,这些路易斯酸/路易斯碱控制剂的使用改变了溶剂的溶解度参数,使反应产物形成一种可变形的复合凝胶结构,其目的为保留足够的端基流动性以使聚合能够继续进行,即,使得聚合物络合物在溶液中保留的时间更长,但仍允许生产高分子量产物所需的聚合物链的流动性。但是这种方法同样有缺陷,这使得更高分子量的聚合物提前析出而停止反应,小分子量的继续增长或溶于反应液中,因此在后期处理过程中将导致分子量分布较宽,以至于形成的聚合物性能难以满足需求。其他缺点包括:由于其极高的堆积密度,需要大量的水或水的酸性溶液才能使聚合物解络合,后处理成本较高;更重要的是,采用该方法得到的反应产物是一种复合凝胶结构,处理聚合物凝胶复合物所需的专用设备需要更高成本。
[0011][0012]
亲电和亲核工艺均会由于副反应而产生使熔体不稳定性的产物。亲核过程中使用的非常高的温度会导致聚合物链的交联,或小分子环状呫吨结构等副反应,从而导致产物中的键的排列达不到理论预期。这两个过程相关的高温也会促进副反应和形成凝胶(可能是交联的颗粒),从而导致最终产品的熔体不稳定。这些凝胶可能显示为变色的杂质物(如黑色物质)。这使得具有精细结构材料(例如纤维和薄膜)的生产变得困难,或者在某些情况下是不可能的。
[0013]
为了促进pekk的亲电合成,溶剂系统可以为美国专利us3956240公开的强酸性溶剂体系(例如hf/bf3),或美国专利us4396755公开的全氟烷基磺酸。然而,这些溶剂系统具有高度腐蚀性,同样存在设备腐蚀的问题。
[0014]
迄今为止,尽管pekk具有优越的性能,但上述问题使得大规模生产高纯度和熔融稳定的pekk成为问题。
[0015]
因此,在本领域中,期待开发一种聚醚酮酮树脂的制备方法,使得聚醚酮酮树脂在可以大规模生产的同时,还具有高纯度、高分子量、并且熔融稳定。
技术实现要素:
[0016]
针对现有技术的不足,本发明的目的在于提供一种通过friedel-crafs亲电取代反应制备聚醚酮酮树脂的方法。本发明的制备方法简单,成本低廉,可大规模生产,同时生产的聚醚酮酮具有高纯度、高分子量以及优越的机械性能。
[0017]
为达此目的,本发明采用以下技术方案:
[0018]
第一方面,本发明提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0019]
(1)将对苯二甲酰氯(tpc)、和/或间苯二甲酰氯(ipc)、封端剂和聚合溶剂在低温下混合,得到混合液;
[0020]
(2)将二苯醚(dpe)、对苯二甲酰氯和聚合溶剂在低温下混合,而后加入lewis酸催化剂,保温,升温,反应,得到反应液;
[0021]
(3)将步骤(2)的反应液降温,而后加入步骤(1)得到的混合液,然后加入lewis酸催化剂,保温,升温,反应,得到反应产物;
[0022]
(4)将步骤(3)的反应产物转移至预先加入有冰的质子溶液的搅拌釜内,后处理,得到所述聚醚酮酮树脂;
[0023]
步骤(2)中二苯醚与对苯二甲酰氯的摩尔比≥2,例如2:1、2.03:1、2.05:1、2.08:1、2.1:1、2.3:1、2.5:1或3:1等,优选2:1。如果二苯醚与对苯二甲酰氯的摩尔比过大,会造成原料的浪费,反应结束后不易将多余的原料除去,也会增加成本和工序。
[0024]
在本发明中,先将过量的二苯醚和部分对苯二甲酰氯反应,反应达到预想的程度后,降低温度,然后补加对苯二甲酰氯和间苯二甲酰氯进行pekk的合成,采用本发明的制备方法,副反应较少,比如有较低的呫吨结构的产生,同时生成的pekk为粒径小于1mm的颗粒或片状,此外,本发明过早析出的聚合物,不具有较低的特性粘度,相反,会具有更高的分子量。
[0025]
在本发明中,只通过加入lewis酸催化剂,不需加入lewis碱催化剂便能实现原料的溶解,并且,制备得到的pekk具有高纯度,高机械性能和较高的熔融稳定性,完全满足特种工程塑料的需求。
[0026]
在本发明的反应体系中,单体二苯醚反应位点有6个,分别在对位和邻位,且对位活性远大于邻位活性,第一段反应过程中,二苯醚的量大于对苯二甲酰氯量,且反应温度控制在低温,从反应热力学分析,对位反应几率远大于邻位反应几率;同时,从立体化学的角度分析,对位位阻远小于邻位的位阻,对位反应几率也远大邻位反应几率,从而很大程度上减少副反应发生的几率,从而使得反应向着预期的方向进行。
[0027]
在本发明中,通过控制步骤(2)中二苯醚与对苯二甲酰氯的摩尔比≥2,可以使得体系内具有更多的1,4-二(苯氧基苯甲酰基)苯(1,4-ekke)结构,这种结构有利于后续形成更为规整的pekk,并且通过控制步骤(2)中二苯醚与对苯二甲酰氯的摩尔比≥2,可以抑制其他副产物的产生,比如可以抑制呫吨结构的杂质产生,而这一杂质结构对pekk的熔体热稳定动性和加工性能具有显著的影响。
[0028]
在本发明中,步骤(2)反应得到的产物为1,4-二(4-苯氧基苯甲酰基)苯(英文缩写为bpbb),其结构如下:
[0029][0030]
在本发明中,步骤(1)所述将对苯二甲酰氯、和/或间苯二甲酰氯、封端剂和聚合溶剂在低温下混合包括三种技术方案,具体地包括以下三种技术方案:
①
将对苯二甲酰氯、封端剂和聚合溶剂在低温下混合;
②
将间苯二甲酰氯、封端剂和聚合溶剂在低温下混合;
③
将对苯二甲酰氯、间苯二甲酰氯、封端剂和聚合溶剂在低温下混合。
[0031]
优选地,步骤(1)和步骤(2)所述低温均为-50℃~-5℃,例如-50℃、-45℃、-40℃、-35℃、-30℃、-25℃、-20℃、-15℃、-10℃或-5℃等,并且步骤(1)和步骤(2)所述低温的温度相同。
[0032]
优选地,步骤(3)所述降温为降至-50℃~-5℃,例如-50℃、-45℃、-40℃、-35℃、-30℃、-25℃、-20℃、-15℃、-10℃或-5℃等。
[0033]
优选地,所述步骤(1)在混合釜中进行,所述步骤(2)和步骤(3)在聚合釜中进行。所述聚合釜、混合釜可为不锈钢釜、搪瓷釜或玻璃釜等。
[0034]
作为本发明的优选技术方案,步骤(1)、步骤(2)所述低温和步骤(3)所述降温后的温度保持一致,这样可以防止混合釜或聚合釜内溶液体系温度过高,而显著地聚合成聚醚酮酮;同时,不用再花费更多时间让温度达到均一。
[0035]
优选地,所述封端剂包括苯甲酰氯、4-苯氧基二苯甲酮、苯磺酰氯,4-氯联苯、双(4-苯氧基苯基)甲酮或联苯中的任意一种或至少两种的组合。若不加入封端剂,在完全化学平衡的情况下,聚醚酮酮具有很高的特性粘度,这使得其难以进行熔融加工,限制了其应用,虽然也可以通过其他的方式(比如溶液法)进行加工,但是pekk本身难以溶于通常的溶剂,需要像浓硫酸等高浓度的酸才能将其进行溶解,但这并不是我们完全所希望的;封端剂终止了该链的至少一端的持续生长从而控制了所得聚合物的分子量,而得到分子量较低的聚合物,从而降低聚合物分子量。因此,使反应计量失衡,即实际的计量为苯二酰氯和二苯醚化学计量为1:1为平衡,通过添加封端剂,使得比例失衡,控制添加不同的量来控制得到具有不同分子量可熔融加工的目标聚醚酮酮聚合物。
[0036]
优选地,以步骤(1)和步骤(2)中对苯二甲酰氯和间苯二甲酰氯的总质量为100%计,所述封端剂的加入量为0.1%-2%,例如0.1%、0.3%、0.5%、1%、1.3%、1.5%、1.8%或2%等。
[0037]
优选地,步骤(1)和步骤(2)所述聚合溶剂各自独立地包括二氯甲烷、1,2-二氯乙烷、二硫化碳、邻二氯苯、间二氯苯、对二氯苯、1,2,3-三氯苯、四氯乙烯、1,2,4-三氯苯、邻二氟苯、硝基甲烷、硝基苯、2-二氯乙烷二氯苯或1,1,2,2-四氯乙烷中的任意一种,优选为邻二氯苯或二氯甲烷,更优选为二氯甲烷。
[0038]
优选地,以步骤(1)和步骤(2)所述对苯二甲酰氯和间苯二甲酰氯的总重量为100%计,所述对苯二甲酰氯的加入量为50-100%,例如50%、60%、70%、80%、90%或100%等,所述间苯二甲酰氯的加入量为0-50%,例如0、10%、20%、30%、40%或50%等。可
根据加工和性能需求,合成不同tpc和ipc比率的pekk,这也是pekk优于同种聚芳醚酮的一点。
[0039]
优选地,步骤(2)中二苯醚的质量浓度为步骤(2)体系内所有物质的2~20%(即,以步骤(2)中加入的二苯醚、对苯二甲酰氯和聚合溶剂的总质量为100%计,二苯醚的加入量为2~20%),例如2%、3%、5%、8%、10%、13%、15%、18%或20%等,对苯二甲酰氯的浓度为步骤(2)体系内所有物质的1~10%,例如1%、2%、3%、5%、8%或10%等。
[0040]
优选地,步骤(2)和步骤(3)所述lewis酸催化剂各自独立地包括无水氯化铝、三溴化铝、五氯化锑、五氟化锑、氯化锢、氯化嫁、氯化棚、氟化棚、氯化铮、氯化铁、氯化锡、四氯化钛或五氯化钼中的任意一种或至少两种的组合,优选无水氯化铝。
[0041]
优选地,步骤(2)所述lewis酸催化剂的加入量为步骤(2)体系内羰基摩尔总量的0.5~4倍,例如0.5倍、1倍、1.5倍、2倍、2.5倍、3倍、3.5倍或4倍等。
[0042]
优选地,步骤(3)所述lewis酸催化剂的加入量为步骤(3)体系内羰基摩尔总量的0.5~4倍,例如0.5倍、1倍、1.5倍、2倍、2.5倍、3倍、3.5倍或4倍等。
[0043]
优选地,步骤(2)所述加入lewis酸催化剂为在5min~360min(例如5min、10min、20min、30min、60min、120min、180min、240min、300min或360min等,优选10min~240min,进一步优选20min~180min,更优选30min~120min)内加入完毕,并且在加入lewis酸催化剂过程中,保持体系温度为-20℃~40℃,例如-20℃、-15℃、-10℃、-5℃、0℃、10℃、20℃、30℃或40℃等,优选-15℃~30℃,进一步优选-10℃~20℃,进一步优选-5℃~10℃。
[0044]
优选地,步骤(2)所述保温的温度为-50℃~-5℃,例如-50℃、-45℃、-40℃、-35℃、-30℃、-25℃、-20℃、-15℃、-10℃或-5℃等,保温的时间为5min~120min,例如5min、10min、20min、30min、60min、90min或120min等,更优选10min~60min。
[0045]
优选地,步骤(2)所述升温为升温至-5℃~150℃,例如-5℃、0℃、10℃、20℃、30℃、40℃、50℃、60℃、70℃、80℃、90℃、100℃、110℃、120℃、130℃、140℃或150℃等,步骤(2)所述反应的时间为20min~360min,例如20min、30min、40min、50min、60min、120min、180min、240min、300min或360min等,优选30min~300min,进一步优选40min~240min,更优选50min~180min。
[0046]
优选地,步骤(3)所述加入lewis酸催化剂为在5min~360min(例如5min、10min、30min、60min、120min、180min、240min、300min或360min等)内加入完毕,并且在加入lewis酸催化剂过程中,保持体系温度为-20℃~40℃,例如-20℃、-15℃、-10℃、-5℃、0℃、10℃、20℃、30℃或40℃等。
[0047]
优选地,步骤(3)所述保温的温度为-50℃~-5℃,例如-50℃、-45℃、-40℃、-35℃、-30℃、-25℃、-20℃、-15℃、-10℃或-5℃等,保温的时间为5min~120min,例如5min、10min、20min、30min、60min、90min或120min等。
[0048]
在本发明中,步骤(2)和步骤(3)中加入lewis酸催化剂后保温一段时间有利于原料混合均匀,体系温度均一,反应缓慢进行,从而使得产物的结构更为均一,同时可以降低副反应的产生。
[0049]
优选地,步骤(3)所述升温为升温至-5℃~150℃,例如-5℃、0℃、10℃、20℃、30℃、40℃、50℃、60℃、70℃、80℃、90℃、100℃、110℃、120℃、130℃、140℃或150℃等,步骤(3)所述反应的时间为20min~360min,例如20min、30min、60min、120min、180min、240min、
300min或360min等。
[0050]
作为本发明的优选技术方案,反应进行的温度可以为-50℃至150℃。优选在较低温度下开始反应,例如可以在-50℃至-5℃下开始反应,特别是体系内包含高反应活性单体时,从低温开始,不仅可以控制反应的速率,还很大程度上减少副反应的产生。聚合开始后,如果需要可以升高温度,例如通过升高温度以提高反应速率。通常优选在-5℃~90℃下进行反应,本发明优先在低温常温下反应。
[0051]
优选地,所述质子溶液包括水、稀盐酸水溶液、稀盐酸甲醇溶液、甲醇、乙醇、丙酮、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二甲醚、乙醚、四氢呋喃、三甲胺、三甲胺盐酸盐、二甲基硫醚、四亚甲基砜、二苯甲酮、四甲基氯化铵或异丙醇中的任意一种或至少两种的组合,优选冰水或冷却的稀盐酸水溶液或稀盐酸甲醇溶液。在本发明中,所述质子溶液作为解络合试剂进行解络合。解络合步骤为剧烈放热反应,因此沸点较低的二氯甲烷、甲醇等,可通过其自身放热使混合液沸腾,从而除去大部分有机溶剂,另外,通过外加冷凝装置加以回收,重复利用,降低成本。本发明所用解络合釜进行解络合时在常压下进行。
[0052]
优选地,所述稀盐酸水溶液或稀盐酸甲醇溶液中,盐酸的重量百分比为0.1~10%,例如0.1%、0.5%、1%、2%、3%、4%、5%、6%、7%、8%、9%或10%等,优选0.5%~5%。
[0053]
优选地,所述质子溶液的加入量大于步骤(2)和步骤(3)中lewis酸催化剂的总量,即,大于反应混合物中存在的结合的(络合的)和未结合的lewis酸催化剂的总量,并且优选大于lewis酸催化剂总量的2倍。
[0054]
优选地,所述冰的质子溶液的温度为-5℃~5℃,例如-5℃、-4℃、-3℃、-2℃、-1℃、0℃、1℃、2℃、3℃、4℃或5℃等。
[0055]
优选地,步骤(4)所述后处理包括固液分离、打碎、清洗和干燥的步骤。
[0056]
优选地,所述打碎聚醚酮酮的设备为胶体磨或锤磨机,所述清洗聚醚酮酮的设备为平板式间歇式离心机或碟片式连续离心机。
[0057]
优选地,为了得到较高纯度的pekk聚合物,需对其进行清洗纯化,所用的清洗剂为水、甲醇、乙醇、丙酮、二氯甲烷、1,2-二氯乙烷、邻二氯苯、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺或n-甲基吡咯烷酮中的一种或至少两种的组合。为了能达到清洗纯化的目的,并且考虑环保问题,优先采用可与水互溶的清洗剂,以便用水置换去除。本发明优选的清洗溶剂为水、甲醇、n,n-二甲基甲酰胺或n-甲基吡咯烷酮等。
[0058]
另外,本发明所用溶剂、清洗剂、解络合剂(质子溶液)都可进行分离纯化后重复利用。
[0059]
作为本发明的优选技术方案,所述制备方法包括以下步骤:
[0060]
(1)将对苯二甲酰氯、和/或间苯二甲酰氯、封端剂和聚合溶剂加入混合釜中,在-50℃~-5℃下混合,得到混合液;
[0061]
(2)将二苯醚、对苯二甲酰氯和聚合溶剂加入聚合釜中,在-50℃~-5℃下混合,而后在5min~360min内将lewis酸催化剂加入完毕,并且在加入lewis酸催化剂过程中保持体系温度为-20℃~40℃,而后在-50℃~-5℃下保温5min~120min,升温至-5℃~150℃,反应20min~360min,得到反应液;
[0062]
(3)将步骤(2)的反应液降温至-50℃~-5℃,而后加入步骤(1)得到的混合液,然
后在5min~360min内将lewis酸催化剂加入完毕,并且在加入lewis酸催化剂过程中保持体系温度为-20℃~40℃,而后在-50℃~-5℃下保温5min~120min,升温至-5℃~150℃,反应20min~360min,得到反应产物;
[0063]
(4)将步骤(3)的反应产物转移至预先加入有冰的质子溶液的搅拌釜内,后处理,得到所述聚醚酮酮树脂;
[0064]
步骤(2)中二苯醚与对苯二甲酰氯的摩尔比≥2。
[0065]
第二方面,本发明提供一种聚醚酮酮树脂,所述聚醚酮酮树脂采用如第一方面所述的制备方法制备得到。
[0066]
优选地,所述聚醚酮酮树脂为颗粒状或雪花片状,且粒径小于≤1mm。因其具有极小的粒径,不需要用特有的粉碎设备,采用目前工业常规的粉碎设备(比如胶体磨,锤磨机等)就能被完美粉碎成粉末;其状态较为蓬松,得益于较小的堆积密度,颗粒为多孔空心结构,因此,内部容易残存较多的有机溶剂,通过传统的过滤方式难以除去,通过离心的方式可以很好除去内部残存的溶剂,不仅如此,离心的方式可以同时进行过滤和洗涤步骤而达到相同的效果。因此,可极大增加设备利用率,降低成本,节约时间,离心机可为平板式间歇离心机或碟片式连续式离心机。
[0067]
优选地,所述聚醚酮酮树脂适用的加工成型方法包括挤塑成型、注塑成型、压塑成型、激光烧结、3d成型、粉末喷涂、溶液加工或熔融纺丝中的任意一种。即,将本发明制备的聚醚酮酮树脂进行加工制备成其他制品时,可通过挤塑成型、注塑成型、压塑成型、激光烧结、3d成型、粉末喷涂、溶液加工、熔融纺丝等加工成型。
[0068]
采用本发明的制备方法合成纯化的pekk聚合物,具有极佳的机械性能、热学性能和生物相容性,不仅可满足特种工程塑料方面的应用,而且可用于人体创伤、颅颌面和脊柱修复替代等生物医用领域。
[0069]
采用本发明的制备方法,通过替换原料,还可以制备聚芳基醚酮(paek),其具有以下结构:
[0070]-ar
1-o-ar
1-(c=o)-ar
2-(c=o)-[0071]
其中,ar1和ar2表示芳香族环基团;具体地,ar1代表1,4-亚苯基;ar2代表1,3-亚苯基、1,4-亚苯基、1,3-亚萘基、1,4-亚萘基、1,5-亚萘基、2,6-亚萘基、4,4'-联亚苯基或稠合芳族基团等,优选1,3-亚苯基、1,4-亚苯基或二者的混合。
[0072][0073]
本发明合成的聚醚酮酮的结构包含式a、式b或二者按比例混合的结构。
[0074]
与现有技术相比,本发明至少具有以下有益效果:
[0075]
(1)在本发明中,先将过量的二苯醚和部分对苯二甲酰氯反应,反应达到预想的程
度后,降低温度,然后补加对苯二甲酰氯和间苯二甲酰氯进行pekk的合成,采用本发明的制备方法,副反应较少,同时生成的pekk为粒径小于1mm的颗粒或片状,便于后期处理,简化单元操作。
[0076]
(2)在本发明中,通过控制步骤(2)中二苯醚与对苯二甲酰氯的摩尔比≥2,可以使得体系内具有更多的1,4-二(苯氧基苯甲酰基)苯(1,4-ekke)结构,这种结构有利于后续形成更为规整的pekk,并且通过控制步骤(2)中二苯醚与对苯二甲酰氯的摩尔比≥2,可以抑制其他副产物的产生,比如可以抑制呫吨结构的杂质产生,而这一杂质结构对pekk的熔体热稳定性和加工性能具有显著的影响。
附图说明
[0077]
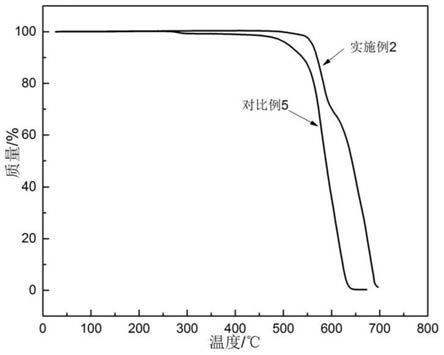
图1为本发明实施例2和对比例5提供的聚醚酮酮树脂的分解温度测试结果图。
具体实施方式
[0078]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0079]
实施例1
[0080]
在本实施例中提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0081]
(1)将20.3g对苯二甲酰氯,0.4g的苯甲酰氯和100ml二氯甲烷加入混合釜中,在-15℃下混合,得到混合液;
[0082]
(2)在经过n2保护下的2l反应釜内加入二苯醚34.04g,对苯二甲酰氯20.3g,500ml的二氯甲烷,将反应体系降温至-15℃,在保持-15℃的情况下30min内缓慢加入42.66g的无水氯化铝,保持30min后,以4℃/min的升温速率升温至常温,保持温度反应1h,得到反应液;
[0083]
(3)将步骤(2)的反应液降温至-15℃,而后加入步骤(1)得到的混合液,然后在30min内通过粉料加料器缓慢加入43.10g的无水氯化铝,并且在加入无水氯化铝过程中保持体系温度低于-5℃,在-15℃下保温1h后,升温至常温反应5h,停止搅拌,得到反应产物;
[0084]
(4)将冰的400g的4%的盐酸水溶液(-2℃)加入搅拌釜内,而后加入步骤(3)的反应产物,在保持体系内温度低于30℃下搅拌30min,将络合氯化铝的聚合物进行解络合,完成后,进行离心固液分离,收集固体,粉碎后,加入600g的n-甲基吡咯烷酮加热回流,再用乙醇回流一次,水洗后,150℃干燥24h,得到所述聚醚酮酮树脂。
[0085]
实施例2
[0086]
在本实施例中提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0087]
(1)将12.18g对苯二甲酰氯,8.12g间苯二甲酰氯,0.4g的苯甲酰氯和100ml二氯甲烷加入混合釜中,在-15℃下混合,得到混合液;
[0088]
(2)在经过n2保护下的2l反应釜内加入二苯醚34.04g,对苯二甲酰氯20.3g,500ml的二氯甲烷,将反应体系降温至-15℃,在保持-15℃的情况下30min内缓慢加入42.66g的无水氯化铝,保持30min后,以4℃/min的升温速率升温至常温,保持温度反应1h,得到反应液;
[0089]
(3)将步骤(2)的反应液降温至-15℃,而后加入步骤(1)得到的混合液,然后在30min内通过粉料加料器缓慢加入43.10g的无水氯化铝,并且在加入无水氯化铝过程中保持体系温度低于-5℃,在-15℃下保温1h后,升温至常温反应5h,停止搅拌,得到反应产物;
[0090]
(4)将冰的400g的4%的盐酸水溶液(-2℃)加入搅拌釜内,而后加入步骤(3)的反应产物,在保持体系内温度低于30℃下搅拌30min,将络合氯化铝的聚合物进行解络合,完成后,进行离心固液分离,收集固体,粉碎后,加入600g的n-甲基吡咯烷酮加热回流,再用乙醇回流一次,水洗后,150℃干燥24h,得到所述聚醚酮酮树脂。
[0091]
实施例3
[0092]
本实施例与实施例2的不同之处仅在于,步骤(2)加入的二苯醚量增大,具体地,步骤(2)中在经过n2保护下的2l反应釜内加入二苯醚34.5g,其他步骤均和实施例2相同。
[0093]
实施例4
[0094]
本实施例与实施例2的不同之处仅在于,补加的苯二酰氯的比例改变,具体地,步骤(1)中将4.06g的对苯二甲酰氯,16.25g的间苯二甲酰氯和100ml二氯甲烷加入混合釜中,其他步骤均和实施例2相同。
[0095]
实施例5
[0096]
本实施例与实施例2的不同之处仅在于,补加的苯二酰氯的比例改变,具体地,步骤(1)中将20.3g的间苯二甲酰氯和100ml二氯甲烷加入混合釜中,其他步骤均和实施例2相同。
[0097]
实施例6
[0098]
在本实施例中提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0099]
(1)将12.18g对苯二甲酰氯,8.12g间苯二甲酰氯,0.6g苯甲酰氯和100ml二氯甲烷加入混合釜中,在-15℃下混合,得到混合液;
[0100]
(2)在经过n2保护下的2l反应釜内加入二苯醚34.04g,对苯二甲酰氯20.3g,500ml的二氯甲烷,将反应体系降温至-15℃,在保持-15℃的情况下30min内缓慢加入42.66g的氯化铝,保持30min后,以4℃/min的升温速率升温至常温,保持温度反应1h,得到反应液;
[0101]
(3)将步骤(2)的反应液降温至-15℃,而后加入步骤(1)得到的混合液,然后在30min内通过粉料加料器缓慢加入43.73g的无水氯化铝,并且在加入无水氯化铝过程中保持体系温度低于-5℃,保持-15℃反应1h后,升温至常温反应5h,停止搅拌,得到反应产物;
[0102]
(4)将冰的400g的4%的盐酸水溶液(-2℃)加入搅拌釜内,而后加入步骤(3)的反应产物,在保持体系内温度低于30℃下搅拌30min,将络合氯化铝的聚合物进行解络合,完成后,进行离心固液分离,收集固体,粉碎后,加入600g的n-甲基吡咯烷酮加热回流,再用乙醇回流一次,水洗后,150℃干燥24h,得到所述聚醚酮酮树脂。
[0103]
对比例1
[0104]
在本对比例中提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0105]
(1)将12.18g对苯二甲酰氯,8.12g间苯二甲酰氯,0.4g苯甲酰氯和100ml二氯甲烷加入混合釜中,在-15℃下混合,得到混合液;
[0106]
(2)在经过n2保护下的2l反应釜内加入二苯醚33g,对苯二甲酰氯20.3g,500ml的二氯甲烷,将反应体系降温至-15℃,在保持-15℃的情况下30min内缓慢加入42.66g的氯化铝,保持30min后,以4℃/min的升温速率升温至常温,保持温度反应1h,得到反应液;
[0107]
(3)将步骤(2)的反应液降温至-15℃,而后加入步骤(1)得到的混合液,然后在30min内通过粉料加料器缓慢加入43.1g的无水氯化铝,并且在加入无水氯化铝过程中保持体系温度低于-5℃,保持-15℃反应1h后,升温至常温反应5h,停止搅拌,得到反应产物;
[0108]
(4)将冰的400g的4%的盐酸水溶液(-2℃)加入搅拌釜内,而后加入步骤(3)的反应产物,在保持体系内温度低于30℃下搅拌30min,将络合氯化铝的聚合物进行解络合,完成后,进行离心固液分离,收集固体,粉碎后,加入600g的n-甲基吡咯烷酮加热回流,再用乙醇回流一次,水洗后,150℃干燥24h,得到所述聚醚酮酮树脂。
[0109]
对比例2
[0110]
在本对比例中提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0111]
(1)将12.18g对苯二甲酰氯,8.12g间苯二甲酰氯和100ml二氯甲烷加入混合釜中,在-15℃下混合,得到混合液;
[0112]
(2)在经过n2保护下的2l反应釜内加入二苯醚34.04g,对苯二甲酰氯20.3g,500ml的二氯甲烷,将反应体系降温至-15℃,在保持-15℃的情况下30min内缓慢加入42.66g的无水氯化铝,保持30min后,以4℃/min的升温速率升温至常温,保持温度反应1h,得到反应液;
[0113]
(3)将步骤(2)的反应液降温至-15℃,而后加入步骤(1)得到的混合液,然后在30min内通过粉料加料器缓慢加入43.10g的无水氯化铝,保持温度低于-5℃,在-15℃下保温1h后,升温至常温反应5h,停止搅拌,得到反应产物;
[0114]
(4)将冰的400g的4%的盐酸水溶液(-2℃)加入搅拌釜内,而后加入步骤(3)的反应产物,在保持体系内温度低于30℃下搅拌30min,将络合氯化铝的聚合物进行解络合,完成后,进行离心固液分离,收集固体,粉碎后,加入600g的n-甲基吡咯烷酮加热回流,再用乙醇回流一次,水洗后,150℃干燥24h,得到所述聚醚酮酮树脂。
[0115]
对比例3
[0116]
本对比例与实施例2的区别仅在于,步骤(1)和步骤(2)所用的聚合溶剂均为1,2-二氯乙烷,其他步骤均与实施例2相同。
[0117]
对比例4
[0118]
本对比例与实施例2的区别仅在于,步骤(2)和步骤(3)加入无水氯化铝后不经过保温过程,立即升温进行反应,其他步骤均与实施例2相同。
[0119]
对比例5
[0120]
在本对比例中提供一种聚醚酮酮树脂的制备方法,所述制备方法包括以下步骤:
[0121]
(1)在经过n2保护下的2l反应釜内加入二苯醚34.04g,对苯二甲酰氯32.48g,8.12g间苯二甲酰氯,0.4g的苯甲酰氯和600ml的二氯甲烷,将反应体系降温至-15℃,在保持-15℃的情况下30min内缓慢加入85.76g的无水氯化铝,保持30min后,以4℃/min的升温速率升温至常温,保持温度反应6h,停止搅拌,得到反应产物;
[0122]
(2)将冰的400g的4%的盐酸水溶液(-2℃)加入搅拌釜内,而后加入步骤(1)的反应产物,在保持体系内温度低于30℃下搅拌30min,将络合氯化铝的聚合物进行解络合,完成后,进行离心固液分离,收集固体,粉碎后,加入600g的n-甲基吡咯烷酮加热回流,再用乙醇回流一次,水洗后,150℃干燥24h,得到所述聚醚酮酮树脂。
[0123]
对实施例以及对比例制备的聚醚酮酮树脂进行性能测试,测试方法如下:
[0124]
(1)特性粘度:按照iso 307的方法测试聚醚酮酮树脂在96%硫酸中的特性粘度;
[0125]
(2)分解温度:采用热重分析仪,在动态氮气气氛下测试分解温度,升温区间为30℃到700℃,升温速率为10k/min;
[0126]
(3)拉伸强度和断裂伸长率:按照iso527 1ba型压塑制样并按照iso527进行拉伸
强度和断裂伸长率测试。
[0127]
性能测试结果如表1所示,实施例2和对比例5提供的聚醚酮酮树脂的分解温度测试结果图如图1所示。
[0128]
表1
[0129] 特性粘度(dl/g)分解温度(℃)拉伸强度/mpa断裂伸长率/%实施例11.9455414111实施例21.7355213023实施例31.8555312235实施例41.55548106168实施例51.6155198no break实施例60.9554912046对比例10.34521626对比例22.755561359对比例30.8155211317对比例40.7655111811对比例50.685288721
[0130]
由表1可以看出,本发明实施例制备的聚醚酮酮树脂具有合适的特性粘度(0.95-1.94dl/g),优异的熔融稳定性(分解温度:548-554℃)以及较好的机械性能,并且,不同的对苯二甲酰氯和间苯二甲酰氯比率,使得聚醚酮酮树脂的拉伸强度不同,对苯二甲酰氯含量越高,聚醚酮酮树脂的拉伸强度越大。
[0131]
相比于实施例2,对比例1、对比例3-4提供的聚醚酮酮树脂的特性粘度和拉伸强度均明显下降,虽然对比例2提供的聚醚酮酮树脂的分解温度和拉伸强度稍有提升,但由于其特性粘度过大,导致聚醚酮酮树脂的加工性能变差。
[0132]
相比于实施例1-6,对比例5提供的聚醚酮酮树脂的特性粘度和拉伸强度均明显下降,分解温度也有所下降。
[0133]
此外,从图1中可以看出,相比于对比例5,实施例2提供的聚醚酮酮树脂具有较高的分解温度,并且熔融稳定性更好。
[0134]
因此,采用本发明的技术方案,使得聚醚酮酮树脂在可以大规模生产的同时,还具有高纯度、高分子量、并且熔融稳定。
[0135]
申请人声明,本发明通过上述实施例来说明本发明的聚醚酮酮树脂的制备方法,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1