一种荧光分析检测APE1酶的三维DNAwalker生物传感器
一种荧光分析检测ape1酶的三维dnawalker生物传感器
技术领域
1.本发明属于生物化学分析方法,具体涉及一种荧光分析检测ape1酶的三维多足dna walker生物传感器。
背景技术:
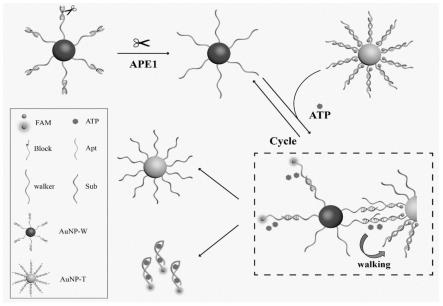
2.脱嘌呤/脱嘧啶核酸内切酶又称氧化还原效应因子1(即ape1)是一种重要的dna修复蛋白,在大多数肿瘤细胞中过度表达,被认为是癌症风险评估、诊断、预后和治疗效果的标志物。尽管ape1在癌症诊断方面有重要意义,但在活细胞中感知ape1酶的丰度变化、监测酶活性仍然是一项挑战。dna walker的实现源于dna出色的可编程性,dna walker的渐进运动提供了执行多项任务和信号放大的可能性,为分子运输,生物传感器和生物合成提供了有价值的平台。在已开发的dna walker的体系中,有不同的驱动力推动dna walker的渐进,比如核酸酶,dnazyme,链置换作用或者少数特殊作用,但均需要添加一些外源性辅助因子。而atp是大多数细胞过程的主要能量来源,并且普遍存在于每个细胞的细胞质和核质中,作为一种内源性驱动力,不需要任何辅助作用,就可以驱动dna walker的有效信号放大,为细胞内ape1的成像奠定一定的优势,结合灵敏的荧光分析方法,为ape1酶的检测与成像提供了一种独特的思路。
技术实现要素:
3.有鉴于此,本发明提供一种荧光分析检测ape1酶的三维多足dna walker生物传感器。具体提供了如下的技术方案:
4.1、一种荧光分析检测ape1酶的三维多足dna walker生物传感器,步骤为:
5.1)用正丁醇法,以巯基dna-walker与aunps摩尔比为500:1的比例修饰aunps,离心清洗三次,溶于超纯水中得到aunp-w;
6.2)用正丁醇法,以巯基dna-sub与aunps摩尔比为1000:1的比例修饰aunps,离心清洗三次,溶于纯水中得到aunp-t;
7.3)用tae/mg
2+
缓冲液将步骤1)得到的aunp-w与封闭链block退火形成双链aunp-w/b;
8.4)用tae/mg
2+
缓冲液将步骤2)得到的aunp-t与atp适体链apt(含fam荧光团)退火形成双链aunp-s/a;
9.5)将步骤3)4)得到的aunp-w/b、aunp-s/a与atp混合得到荧光分析检测ape1酶的生物传感器;
10.6)将不同浓度的ape1酶加入荧光分析检测ape1酶的生物传感器中,放入rt-qpcr中37℃进行反应,测得不同浓的ape1酶的荧光曲线;
11.7)把待测样品加入步骤5)得到荧光分析检测ape1酶的生物传感器中,37℃下孵育,设定与步骤6)一样的检测条件,测得不同待测样品的荧光曲线,获得ape1酶的特异性检测结果。
12.进一步,步骤6)所述的双金球aunp-w/b、aunp-s/a、ape1酶与atp 37℃反应的时间为1h
–
3h。
13.进一步,步骤6)所述的双金球aunp-w/b、aunp-s/a、ape1酶与atp 37℃反应的时间为3h。
14.进一步,所述的巯基修饰的dna walker序列为seq id no.1;所述的封闭链block序列为seq id no.2;所述的atp适体链apt的序列为seq id no.4;所述的dna sub序列为seq id no.6。
15.进一步,步骤1)所述的tae/mg
2+
缓冲液组成为40mm tris,20mm醋酸,1mm edta,10mm mg
2+
,ph=8。
16.进一步,步骤1)所述的正丁醇法修饰aunps,在200μl体系中,加入1.8ml的正丁醇,快速涡流搅拌混合均匀,加入0.4ml的0.5
×
tbe,快速涡流搅拌混合均匀,在2000g的离心力下离心,清洗三次,产物溶于超纯水中。
17.进一步,步骤3)所述的aunp-w与封闭链block杂交,block链过量1.2倍;所述的aunp-t和apt等摩尔量。
18.进一步,步骤5)所述的atp的用量为1-10mm。
19.进一步,步骤5)所述的atp的用量为1mm。
20.进一步,步骤5)所述的aunp-w/b、aunp-s/a的比例为1:4。
21.进一步,步骤6)所述的ape1酶范围为0.1-1000u/ml。
22.进一步,步骤6)所述的rt-qpcr的参数设置为temperature:37℃,acquisition:green。
23.本发明的有益效果在于:基于aunps有良好的生物相容性并且具有猝灭荧光的作用,结合三维dna walker高效、稳定的优点,构建了一种由atp驱动dna walker信号放大的生物传感器用于ape1酶的荧光检测。基于荧光快速响应的优点,设计了由两种金球组成的dna walker体系,其中aunp-w充当多足dna walker载体,另一种aunp-t充当walker行走轨道的作用,可以实现aunp-w围绕aunp-t行走的效果。在识别靶标之前,我们将block整合到aunp-w中以封闭所有的walker链,另外,将atp适配体apt(含fam荧光团)杂交到aunp-t上,fam靠近aunps,发生fret效应使得fam荧光被猝灭。当识别靶标ape1时,block/walker双链中的ap位点被切割,解锁多足walker(aunp-w),与aunp-t上的apt发生反应,随后在atp的辅助作用下,重新释放walker,进行下一步行走反应。在这个过程中,atp与适配体的结合作用充当dna walker持续行走的动力,由于链置换的作用,apt上的fam基团已经远离aunps,fret效应消失,荧光获得恢复。在整个实验方案中,通过atp驱动dna walker,结合fret效应实现荧光由猝灭到恢复的策略,可用于ape1酶的检测。当靶标ape1酶不存在的时候,block/walker双链中的ap位点不会被切割,无法引发后续反应,则不会产生荧光恢复信号。本发明将dna walker信号放大作用与荧光分析结合,加入内源性atp作为持续行走的驱动力,ape1酶作为walker启动的触发开关,过程简单易行、响应迅速、成功的用于ape1酶的检测,有望在细胞中监测ape1酶活性,为下一步在细胞内成像奠定了体外检测基础,具有潜在的应用价值。
(seq id no.6);
45.sub-1序列为5
’‑
sh-ttt ttt ttt tcc tcc gct ttt ttc ccc cag gt-3’(seq id no.7);
46.sub-2序列为5
’‑
ttt ttt tta acc tcc gct ttt ttc ccc cag gt-3’(seq id no.8);
47.实施例2聚丙烯酰胺凝胶电泳进行方案可行性验证
48.首先,对block与walker进行退火处理,以形成稳定的双链结构,然后对双链进行酶切处理,10%page结果如图2所示。图2中条带1、2分别表示block单链与walker单链;条带3表示block/walker杂交双链结构,大概位于50bp的位置;条带4验证了walker的背景信号值,即在block/walker双链与apt共同存在时,37℃反应1h,条带仍然显示出明显的block/walker双链与apt单链条带,表明walker可以很好的与block杂交,不会因为apt的存在产生假阳性信号。随后,条带5在block/walker双链基础上加入ape1酶,37℃孵育1h后,我们观察到block/walker双链完全消失,全部变为walker链,其位置低于50bp,证明了ape1酶成功的切割了block/walker双链中的ap位点,block脱离walker变为两条短链,释放所有的walker链可以用于后续实验;最后,我们在条带6中,证实了walker能够与apt结合是ape1酶存在的结果,将block/walker双链、ape1酶与apt单链混合反应,block/walker双链与apt单链均消失,在接近60bp的位置出现walker/apt双链结构,表明ape1酶切后,block无法继续与walker结合,释放的walker与apt结合了。综上所述,page的结果表明我们的靶标ape1可以识别双链中ap位点,释放的walker可以进行下一步反应。聚丙烯酰胺凝胶电泳的结果验证了我们方案的可行性。
49.实施例3atp浓度优化
50.实验发现,在atp浓度过高时,sub与apt的结合并不稳定,这是由于sub与apt中间留有5个不互补碱基,其作用是使walker能够更好的置换apt。为了降低sub与apt的背景信号,通过对atp浓度进行优化以降低apt直接被atp带走的可能性。在sub与apt先杂交形成双链的前提下,分别加入不同浓度的atp,10%page结果显示如图3,在10mm的高浓度atp存在时,sub/apt双链极不稳定,大部分sub/apt双链已经解开,仅有少部分双链的存在,并且在20bp处产生大量的apt链,说明10mm的atp条件下,双链是不能够完全稳定存在的。于是依次降低atp浓度,从7.5mm的atp开始,已经转变为sub/apt双链存在更多,apt单链减少。结合图3,可以发现随着atp浓度降低,sub/apt双链逐渐增多,而单链apt强度逐渐减少,到2.5mm的atp之后,已经可以忽略被atp结合的apt链,再继续降低atp浓度,对体系的影响变小。
51.实施例4apt序列优化
52.调整sub/apt双链与atp结合的背景信号之后,需要去平衡walker置换apt的能力,因此我们对apt的序列结构进行了优化。其中apt-0(序列为seq id no.3)是末端有5个碱基暴露的杂交域,杂交域的作用是能够引发与walker的结合;而apt-1(序列为seq id no.4)在apt-0的基础上,在5’末端增加一个杂交的碱基,即6个可以与walker结合的碱基;apt-2(序列为seq id no.5)则是在apt-0的5’端增加两个杂交的碱基,即7个可以与walker结合的碱基。
53.apt序列的优化结果如图4的page所示,条带1至条带3表示了walker/apt结合后,能够被atp置换下来的效率,在我们的体系中,atp作为驱动walker步行的动力,我们希望
walker/apt双链中apt尽可能的被atp结合,从而可以重复释放walker,进行循环行走。随着apt序列的变化,apt与walker的结合能力逐渐增强,当apt由apt-0变为apt-2时,能够被atp结合走的apt链减少,walker/apt双链大量存在,这是我们不希望的结果,因此我们选择apt-0或者apt-1去参与后续反应。条带4、5、6则验证了在相同浓度walker的存在下,walker能否置换出sub/apt双链中的apt,图中结果显示随着apt与walker结合碱基个数增加,apt更容易被walker置换出来。条带7至条带9分别表示了在相同atp浓度下,sub/apt-0、sub/apt-1与sub/apt-2的背景信号大小,我们发现sub/apt-0、sub/apt-1与sub/apt-2的背景信号差异不大,因为这一部分关键还是取决于sub与apt结合的碱基是相同的。最后,条带10-11,则验证了walker、sub/apt、atp共同混合下,37℃孵育,最终结果表明apt-1既有低的背景信号,又能够使walker/apt-1被atp驱动,还能够使得walker置换sub/apt-1中的apt-1,最终我们选择序列apt-1(记为apt)去参与后续的荧光分析实验。
54.实施例5sub序列优化
55.前文中已经优化了atp的浓度以降低背景信号,但是由于荧光测量的灵敏性,在rt-qpcr中,仍然显示出一定的背景信号荧光值,我们需要进一步通过优化sub的序列以增强sub与apt的结合力,降低背景信号的荧光值,并且希望优化的sub序列不影响walker置换apt的能力。因此,我们在不同的sub序列下分别测量了实验组即完整的aunp-w与aunp-t在ape1以及atp同时作用下的荧光曲线,以及对照组(control组)即aunp-w与aunp-t在仅有atp作用下的的荧光曲线。我们进行了三种sub序列的对比,分别为sub-2(7nt,序列为seq id no.8)、sub-1(9nt,序列为seq id no.7)、sub-0(10nt,序列为seq id no.6),分别指的是在5’侧能够与apt杂交的碱基个数,。
56.sub序列的优化结果如图5所示,显示了三种sub序列的实验组与对照组的实时荧光定量曲线,实验结果显示,在180min时,三种sub序列的实验组荧光可以达到一致,但是仅有atp存在时的对照组荧光出现差异,我们可以观察到随着sub与apt杂交个数的增加,其杂交结合能力更强,相对应的背景荧光会降低,sub-0(10nt)的背景荧光最低。因此,在接下来的实验中,我们均使用sub-0(10nt)进行之后的反应,记为sub链。
57.实施例6rt-qpcr实时监测荧光分析验证实验可行性
58.为了验证atp驱动3d dna walker实验的可行性,我们通过rt-qpcr实时监控整个体系的荧光变化。实验结果在图6中,我们采用aunp-w与aunp-t的比例为1:4,1mm的atp,分别验证了在ape1与atp共同存在、仅ape1存在、仅atp存在、以及不加任何物质的空白对照的情况。荧光实时定量曲线表明仅有aunp-w与aunp-t的空白对照几乎不显示荧光(图6-d),这是由于fam完全被aunps猝灭;当加入ape1或atp时,荧光会有少量的恢复(图6-b、c),这是由于ape1切割block的ap位点后,walker置换了一部分的apt-fam链,远离金纳米粒子后,荧光获得了部分恢复,而加入atp的体系则是由于atp的作用使得少部分sub/apt解开了,少部分的apt-fam被atp带到溶液相中,获得了荧光恢复;但是当ape1与atp同时加入时(图6-a),荧光完全恢复,且刚开始恢复速率快,随后曲线逐渐变缓,这表明我们的靶标ape1识别了ap位点,block断裂后释放walker链,walker去置换apt-fam的过程中,在atp的驱动作用下,实现了多足walker的行走,并将大量的apt-fam链释放到溶液中,证明了我们方案的可行性,荧光量与靶标量呈正相关的关系。
59.实施例7dna walker生物传感器的性能分析
60.如图7所示,在相同条件下,分别对0u/ml、0.1u/ml、1u/ml、10u/ml、100u/ml、500u/ml、1000u/ml的ape1进行了分析。在0-1000u/ml靶标范围内,我们可以观察到随时间的延长,aunp-w在不停的运动,荧光逐渐增强。另外,随着待测物ape1酶浓度的增加,荧光值也逐渐增加。实验结果直观的显示了不同浓度ape1对应的实时定量荧光曲线信号。图8分析了在210min时不同靶标浓度对应的荧光值,其中图8中的插图是在0-1000u/ml范围内荧光信号与靶标浓度关系的线性图,按照标准计算方法(lod=3σ/s,σ为标准偏差,s为斜率),得出的检测限为0.033u/ml(图8插图)
61.实施例8实验选择性分析
62.在实验的反应过程中,将ape1酶换成100u/ml的udg酶、t5 exo酶、lambda exo、exoⅰ,反应3h后,通过荧光分光光度计对其进行荧光测量,结果如图9所示,3d dna walker仍对ape1酶有较高的响应,其余不同种类酶对整个体系的影响很小,证实了本发明有良好的特异性。
63.实施例9实际样品分析
64.借助癌细胞与正常细胞两种细胞裂解液进行探究,一个是癌细胞-hela细胞裂解液(5%),另一个为正常细胞-l929细胞裂解液(5%)。在实验的反应过程中,我们将ape1酶换成5%细胞裂解液,并且在相同条件下进行反应,对210min时的反应体系进行荧光测量,图10的实验结果显示hela细胞裂解液所测得的荧光值相较于正常细胞l929裂解液的要高很多,这是因为hela细胞中ape1的表达会过量,结果表明atp驱动3d dna walker的荧光生物传感器在体外可以区分不同细胞的裂解液,证实了我们设计的荧光方案可以有效的区分癌细胞,对之后在细胞内成像实验起到重要作用。
65.最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1