一种2,4,5三氟苄基溴和2,4,5-三氟苯甲酸的制备方法与流程
1.本发明涉及有机物合成制药技术领域,更具体地说,尤其涉及一种2,4,5三氟苄基溴和2,4,5-三氟苯甲酸的制备方法。
背景技术:
2.日本制药公司盐野义制药公司研发的ensitrelvir(s-217622) 是一种新型治疗新冠病毒的口服药。通过以428名12岁以上轻症状与中度症状感染者为对象实施的临床试验结果显示,在服用了3天的口服药之后,在第四天,有80%的人已经检测不出新冠病毒。服用5 天之后的第6天检测,100%的人身上的新冠病毒已经完全消失,效果十分理想。
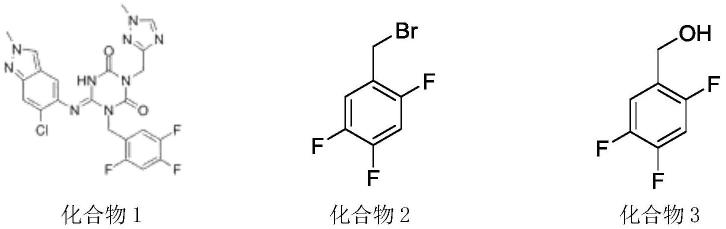
3.2,4,5-三氟苄基溴是合成ensitrelvir的主要结构片段之一,ensitrelvir结构式(化合物1)、2,4,5-三氟苄基溴结构式(化合物2)如下式:
[0004][0005]
2,4,5-三氟苄基溴主要是由2,4,5-三氟苯甲醇(化合物3)溴化获得,2,4,5-三氟苯甲醇是一种重要的医药中间体,工业化生产中主要通过1,2,4-三氟苯氯甲基化反应得到2,4,5-三氟苄基氯,再水解得到2,4,5-三氟苯甲醇,其合成路线如下:
[0006][0007]
2,4,5-三氟苯甲醇中主要存在以下两个杂质(化合物4、化合物5),由于结构相似,后续精制过程中难以去除,并对合成 ensitrelvir产生影响,生产相对应的杂质,导致副作用增加。
[0008]
技术实现要素:
[0009]
针对现有技术的上述缺陷和问题,本发明提供一种2,4,5三氟苄基溴和2,4,5-三氟苯甲酸的制备方法。
[0010]
为了达到上述目的,本发明提供如下技术方案:
[0011]
一种2,4,5三氟苄基溴和2,4,5-三氟苯甲酸的制备方法,包括步骤:
[0012]
步骤一、将2,4-二氯氟苯为原料,通过硝化反应制备得到化合物7;
[0013]
步骤二、将化合物7通过氟化试剂置换得到化合物8;
[0014]
步骤三、化合物8,还原得到化合物9;
[0015]
步骤四、化合物9重氮化反应得到化合物10;
[0016]
步骤五、化合物10通过格氏反应得到化合物3,溴化得到2,4,5
‑ꢀ
三氟苄基溴。
[0017]
上述技术方案中,所述步骤一中,将65%浓硝酸和98%浓硫酸,搅拌升温至50℃,缓慢滴加2,4-二氯氟苯,保温反应2h,停止搅拌,冷却至完全分层,分离有机层,用碳酸氢钠溶液洗涤,55℃减压蒸馏,冷凝馏分,得化合物7。
[0018]
上述技术方案中,所述步骤二中,将化合物7、氟化试剂置于dmso 中,搅拌均匀,升温至80℃,保温反应至主原料反应完毕,冷却至室温,用碳酸钾溶液水洗,分层,60℃减压蒸馏,即得化合物8。
[0019]
上述技术方案中,所述步骤三中,将化合物8、乙醇、钯碳加入加氢釜中,通入氢气,升温至50℃,0.2mpa,加氢反应14h,反应毕,加dmf搅拌均匀,过滤,滤液用碳酸氢钠溶液洗涤,减压浓缩至小体积,降温至10℃保温析晶,过滤,滤饼干燥得化合物9。
[0020]
上述技术方案中,所述步骤四中,将化合物9、溴化氢置水中搅拌均匀,称取硝酸钠置水中,搅拌均匀,将两者重氮化反应,反应温度10-15℃,反应毕,加dmf溶液萃取,合并有机相,减压浓缩至小体积,升温至90℃减压浓缩至无馏分,得化合物10。
[0021]
上述技术方案中,所述步骤五中,将氮气保护下,格氏反应中将镁源加入无水乙醚中,用化合物10与无水乙醚的混合溶液缓慢滴入,滴毕,通入由多聚甲醛解聚成的甲醛气,反应毕,加二氯甲烷搅拌均匀后,用碳酸氢钠溶液洗涤,有机相用无水硫酸钠干燥后,浓缩至小体积,析晶得化合物3;将化合物3置于二氯甲烷中,升温至50℃,通入溴化氢气体至主物料反应完毕,降温至室温,用碳酸氢钠溶液洗涤,有机相用无水硫酸钠干燥后,浓缩至小体积,析晶得2,4,5-三氟苄基溴。
[0022]
上述技术方案中,所述硝化反应的替换方案为采用硝酸银、氯化氢和浓硫酸共同反应制备得到。
[0023]
上述技术方案中,所述步骤二中,氟化试剂包括氟化钾、氟化钠或四丁基氟化铵一种。
[0024]
上述技术方案中,所述步骤四中,重氮化反应采用微通道方式进行。
[0025]
上述技术方案中,所述步骤五中,所述格氏反应采用的镁源为镁屑或异丙基氯化镁。
[0026]
本发明还公开一种2,4,5-三氟苯甲酸的制备方法,其特征在于将上述技术方案中制备得到的化合物3的羟基羧基化,得到高纯度 2,4,5-三氟苯甲酸。
[0027]
本发明使用的起始物料2,4-二氯氟苯价格便宜,其2个氯之间具有较大空间位阻,可有效减少杂质的产生,得到化合物7,后续操作反应条件温和,反应位点单一,生产出的2,
4,5-三氟苄基溴具有较高的纯度,该路线适宜于医药中间体的生产,具有原料药价格易得,操作简单,条件温和,合成收率高,纯度高等优点,具有较高的工业化应用价值。
附图说明
[0028]
图1是本发明实施例1所得到的2,4,5-三氟苄基溴的质谱图谱示意图。
[0029]
图2是本发明实施例2所得到的2,4,5-三氟苄基溴的质谱图谱示意图。
具体实施方式
[0030]
下面将结合本发明的实施例和附图,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0031]
一种2,4,5三氟苄基溴和2,4,5-三氟苯甲酸的制备方法,包括步骤为:以2,4-二氯氟苯为原料,通过硝化反应制备得到化合物7,通过氟化钾置换得到化合物8,还原得到化合物9,重氮化反应得到化合物10,再通过格氏反应得到化合物3,溴化得到2,4,5-三氟苄基溴。反应方程式如下式:
[0032][0033]
上述合成路线中,化合物3即为2,4,5-三氟苯甲醇,是一种重要的化工原料,本发明路线也是一种成熟的制备2,4,5-三氟苯甲醇的方法,制备得到的产品具有纯度高,无化合物4和化合物5杂质的特点。
[0034]
上述合成路线中制备得到的化合物3,通过将羟基羧基化可以制备得到的2,4,5-三氟苯甲酸,2,4,5-三氟苯甲酸是一种精细有机氟合成中间体,可用于特马沙星及液晶等的合成。反应式见下式:
[0035]
[0036]
具体实施方式:
[0037]
实施例1:
[0038]
将浓度为65%浓硝酸50ml和浓度为98%浓硫酸100ml,搅拌升温至50℃,缓慢滴加2,4-二氯氟苯20g后,保温反应2h,停止搅拌,冷却至完全分层,分离有机层,用碳酸氢钠溶液洗涤3次,55℃减压蒸馏,冷凝馏分,得19.8g化合物7。
[0039]
将19.8g化合物7、50g无水四丁基氟化铵置于150mldmso中,搅拌均匀,升温至80℃,保温反应至主原料反应完毕,冷却至室温,用碳酸钾溶液水洗2次,分层,60℃减压蒸馏,即得16.4g化合物8。
[0040]
将16.4g化合物8、80ml乙醇、钯碳加入加氢釜中,通入氢气,升温至50℃,0.2mpa,加氢反应14h,反应毕,加100mldmf搅拌均匀,过滤,滤液用碳酸氢钠溶液洗涤2次,减压浓缩至小体积,降温至10℃保温析晶,过滤,滤饼干燥得13.3g化合物9。
[0041]
将13.3g化合物9、10g溴化氢置40ml水中搅拌均匀,称取15g 硝酸钠置50ml水中,搅拌均匀,将两者置于微通道反应器中,反应温度10-15℃,反应毕,加50mldmf溶液萃取2次,合并有机相,减压浓缩至小体积,升温至90℃减压浓缩至无馏分,得12.1g化合物 10。
[0042]
将氮气保护下,5g的镁屑加入40ml无水乙醚中,用12.1g化合物10与15ml无水乙醚的混合溶液缓慢滴入,滴毕,通入由多聚甲醛解聚成的甲醛气,反应毕,加80ml二氯甲烷搅拌均匀后,用碳酸氢钠溶液洗涤2次,有机相用无水硫酸钠干燥后,浓缩至小体积,析晶得10.1g化合物3。
[0043]
将10.1g化合物3置于80ml二氯甲烷中,升温至50℃,通入溴化氢气体至主物料反应完毕,降温至室温,用碳酸氢钠溶液洗涤2次,有机相用无水硫酸钠干燥后,浓缩至小体积,析晶得9.6g化合物2。
[0044]
根据实施例1制备得到的2,4,5-三氟苄基溴的cg图谱,如图1 所示以及表一数据如下:
[0045]
表一
[0046][0047][0048]
由上表可以看出,用本实施例方法生产的2,4,5-三氟苄基溴纯度达到约99.9%,
具有较高的cg纯度。
[0049]
实施例2:
[0050]
将硝酸银10g、2,4-二氯氟苯20g与浓硫酸70ml,搅拌升温至 40℃,缓慢通入氯化氢气体,控温50℃以下,至主物料反应完毕,停止搅拌,冷却至完全分层,分离有机层,用碳酸氢钠溶液洗涤3次, 55℃减压蒸馏,冷凝馏分,得20.1g化合物7。
[0051]
将20.1g化合物7、20g氟化钾置于100ml乙酸乙酯中,搅拌均匀,升温至80℃,保温反应至主原料反应完毕,冷却至室温,用碳酸钾溶液水洗2次,分层,60℃减压蒸馏,即得17.5g化合物8。
[0052]
将17.5g化合物8、80ml乙醇、钯碳加入加氢釜中,通入氢气,升温至50℃,0.2mpa,加氢反应14h,反应毕,加100mldmf搅拌均匀,过滤,滤液用碳酸氢钠溶液洗涤2次,减压浓缩至小体积,降温至10℃保温析晶,过滤,滤饼干燥得14.6g化合物9。
[0053]
将14.6g化合物9、12g溴化氢置40ml水中搅拌均匀,称取15g 硝酸钠置50ml水中,搅拌均匀,将两者置于微通道反应器中,反应温度10-15℃,反应毕,加50mldmf溶液萃取2次,合并有机相,减压浓缩至小体积,升温至90℃减压浓缩至无馏分,得13.3g化合物 10。
[0054]
将氮气保护下,15g的异丙基氯化镁加入40ml无水乙醚中,搅拌降温至0℃,用13.3g化合物10与15ml无水乙醚的混合溶液缓慢滴入,滴毕,加入8g多聚甲醛,升温至10℃,保温反应10h,反应毕,加90ml二氯甲烷搅拌均匀后,用碳酸氢钠溶液洗涤2次,有机相用无水硫酸钠干燥后,浓缩至小体积,析晶得11.6g化合物3。
[0055]
将11.6g化合物3置于80ml二氯甲烷中,升温至50℃,通入溴化氢气体至主物料反应完毕,降温至室温,用碳酸氢钠溶液洗涤2次,有机相用无水硫酸钠干燥后,浓缩至小体积,析晶得10.2g化合物2。
[0056]
根据实施例1制备得到的2,4,5-三氟苄基溴的cg图谱,如图2 所示以及表二数据如下:
[0057]
表二
[0058]
保留时间峰高峰面积百分比(%)5.0410.34350.0175.5581825.302299.8017.4440.53460.0228.4910.50670.02111.0341.81620.08011.4751.04070.04517.7090.26910.014
[0059]
由上表可以看出,用本实施例方法生产的2,4,5-三氟苄基溴纯度达到99.8%,具有较高的cg纯度。
[0060]
实施例3:
[0061]
2,4,5-三氟苯甲醇的合成:取10g实施例2得到的化合物3溶于 200ml二氯甲烷中,加浓硫酸15ml,搅拌均匀,分次缓慢加入10g高锰酸钾,搅拌保温2h,用碳酸钾溶液洗涤2次,分取有机相,加硫酸钠过夜除水,浓缩至小体积,析晶即得2,4,5-三氟苯甲醇9.1g。实施例3得到的2,4,5-三氟苯甲醇与市售2,4,5-三氟苯甲醇杂质对比情况见表三:
[0062]
表三
[0063][0064]
由上表可以看出,用本发明制备的2,4,5-三氟苯甲醇中未检出杂质1和杂质2且纯度显著高于市售样品1和市售样品2,与现有技术相比具有显著的进步。
[0065]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应所述以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1