一种岩藻多糖酶基因Fcn1及其应用
一种岩藻多糖酶基因fcn1及其应用
技术领域
1.本发明属于基因领域,具体涉及一种岩藻多糖酶基因fcn1及其应用。
背景技术:
2.岩藻多糖是海洋环境中特有的一类多糖,是一类由单糖组成,化学结构都十分复杂的海洋硫酸多糖(10000-100000da),主要来源于褐藻和一些海洋无脊椎动物,具有抗凝血、抗肿瘤、抗血栓、抗病毒、抗氧化等多种生物学功能。然而,岩藻多糖分子结构的复杂性和多样性导致生物功能与其分子结构之间的关系不明确,严重制约了岩藻多糖在临床医药、保健和食品等领域的应用。与高分子量的岩藻多糖相比,低分子量的岩藻寡糖在生物利用度和生物活性方面显示出一定的优势,具有极大的应用潜力。因此,如何通过降解天然大分子量的岩藻多糖获得具有多种生理功能的低分子量岩藻寡糖引起了国内外科研工作者的关注。
3.岩藻多糖酶是降解岩藻多糖的关键酶,由于岩藻多糖种类丰富,因此降解岩藻多糖的菌种和其产生的岩藻多糖酶也各有不同。已报道的在cazy数据库分类的岩藻多糖酶共有18种,岩藻多糖分子结构的复杂性和多样性决定了岩藻多糖酶种类和功能的多样性。目前报道的产岩藻多糖酶菌株存在产酶量低和产出酶的酶活力差的问题,面对上述问题,目前的解决方法主要是,通过优化培养基、培养条件和物理诱变的方式来提升产酶量或者酶活力,但效果并不显著,现在依旧无法实现岩藻多糖酶的商品化,目前关于岩藻多糖酶的研究主要集中在异源表达和最基本的酶学特性表征方面对岩藻多糖酶的相关机理和底物特异性分析都没有更深入的研究。
技术实现要素:
4.本发明的目的在于提供一种岩藻多糖酶基因fcn1及其应用。所述fcn1基因能够编码岩藻多糖酶,其对岩藻聚糖硫酸酯具有高效且专一的降解作用。
5.为实现上述发明目的,本发明采用以下技术方案予以实现:
6.本发明提供了一种岩藻多糖酶基因fcn1,所述fcn1基因的核苷酸序列如seq id no.1所示。
7.进一步的,所述fcn1基因来源于菌株flavobacteriaceae bacterium rc2-3菌株。
8.本发明还提供了用于检测所述的岩藻多糖酶基因fcn1的引物,所述引物的序列为:
9.fcn1-f:5
’‑
ggtaccatgaataaactaatttcaatatttctaggaggg-3’;
10.fcn1-r:5
’‑
ggatccttaatctaaccaagtaattttagcttc-3’。
11.本发明还提供了所述的岩藻多糖酶基因fcn1编码的岩藻多糖酶,所述岩藻多糖酶的氨基酸序列如seq id no.2所示。
12.本发明还提供了含有所述的岩藻多糖酶基因fcn1的重组载体
13.进一步的,所述载体为plb载体。
14.本发明还提供了含有所述的岩藻多糖酶基因fcn1的重组菌株。
15.进一步的,所述重组菌株为大肠杆菌。
16.本发明还提供了所述的岩藻多糖酶基因fcn1或者所述的岩藻多糖酶在用于制备降解岩藻聚糖硫酸酯的制剂中的应用。
17.进一步的,所述岩藻多糖酶基因fcn1编码的岩藻多糖酶通过降解岩藻聚糖硫酸酯中的α1-3糖苷键来达到降解岩藻聚糖硫酸酯的作用。
18.本发明与现有技术相比,具有以下优点和有益效果:
19.1、本发明以已经完成全基因组测序的flavobacteriaceae bacterium rc2-3为研究对象,对其进行基因组生物信息学分析,并与已报道的岩藻多糖酶进行dnaman序列比对,获得一个岩藻多糖酶基因fcn1,其编码的fcn1蛋白(岩藻多糖酶)序列与funa蛋白的同源性达到最近,仅为70.98%。
20.2、本发明获得了fcn1基因的重组菌株,实现了fcn1基因的异源表达,获得异源表达酶岩藻多糖酶,并证明了该酶具有的酶活,且具有降解岩藻聚糖硫酸酯的能力。上述结果不仅丰富了岩藻多糖酶基因家族,为进一步阐明岩藻多糖酶催化作用机制奠定了基础,有助于推动岩藻多糖资源的开发利用,具有重要的理论价值和生产指导意义。
附图说明
21.图1为fcn1与funa蛋白同源序列比对图。
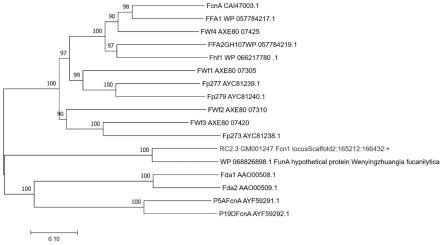
22.图2为fcn1基因编码的氨基酸序列与现有岩藻多糖酶氨基酸序列的亲缘关系图。
23.图3为q5超保真酶pcr扩增基因琼脂糖凝胶电泳结果,其中左边三条带为fcn1基因条带,右边为marker。
24.图4为dna片段进行琼脂糖凝胶电泳回收结果,其中左边条带为fcn1基因条带,右边为marker。
25.图5为含有氨苄霉素抗性lb平板上生长的菌落。
26.图6为阳性转化子琼脂糖凝胶电泳验证图,其中左边条带为单菌落pcr验证条带,右边为marker。
27.图7为质粒酶切结果电泳结果。
28.图8为sds-page凝胶电泳图,其中空白条带为空质粒并进行诱导表达的bl21大肠杆菌上清,1条带到4条带均为转化成功的并且诱导表达了的bl21大肠杆菌,m条带为marker。
具体实施方式
29.结合以下具体实例对本发明的技术方案作进一步详细的说明。
30.下述实施例中,如无特殊说明,所使用的实验方法均为常规方法,所用材料、试剂等均可从生物或化学试剂公司购买。
31.培养基的配置:
32.lb培养基(w/v):0.5%酵母粉,1.0%胰蛋白胨,1.0%nacl,若固体培养基需添加2.0%的琼脂。蒸馏水配制,121℃灭菌20min,冷却至室温后添加终浓度为100.0μg/ml的氨苄青霉素(amp)。
33.tb培养基:
①
胰蛋白胨12g,酵母提取物24g,甘油4ml,加去离子水至900ml,完全溶解,灭菌;
②
kh2po
4 2.31g,k2hpo
4 12.54g,加去离子水至100ml,完全溶解,灭菌。灭菌结束后,在无菌环境下将溶液
①②
混匀。
34.实施例1:fcn1基因的获取
35.从海带中筛选获得一株可以降解岩藻多糖的海洋细菌flavobacteriaceae bacterium rc2-3菌株,对其进行全基因组测序(rc2-3菌株的基因组序列已经提交到genome sequence archive(gsa)数据库,并获得登录号:cra003853),并分析获得结果,利用bioedit软件建立flavobacteriaceae sp.rc2-3菌株基因组数据库和蛋白质数据库。通过ncbi数据库搜素到目前文献中已报道的岩藻多糖酶的基因序列与氨基酸序列记录,目前已报道的岩藻多糖酶信息在表1中可见,将ncbi数据库中已经发布的岩藻多糖酶的基因序列和蛋白序列分别与flavobacteriaceae bacterium rc2-3菌株基因组数据库和蛋白数据库比对,在该菌株中发现了1个岩藻多糖酶基因fcn1,并将flavobacteriaceae bacterium rc2-3菌株中的fcn1基因指导编码的氨基酸序列,与表1中从文献中查找搜寻到的已报道的岩藻多糖酶基因所对应的氨基酸序列,利用mega 7.0软件对其氨基酸序列进行比对,通过neighbor-joining法进行进化树的构建分析。
36.表1已报道的岩藻多糖酶
[0037][0038]
结果如图1-2所示,fcn1基因编码所产的岩藻多糖酶与funa(wp_068826898.1)岩藻多糖酶蛋白序列同源性达到70.98%,并且氨基酸序列系统发育树显示两种酶亲缘关系最近,结合fcn1基因编码的氨基酸序列与funa基因编码的氨基酸序列保守结构域分析结果,fcn1基因编码的蛋白与funa基因编码的蛋白属于同一个糖苷水解酶家族-gh168,这个家族成员不仅可以特异性水解2-o-硫酸化和非硫酸化岩藻糖残基之间的α-1,3糖苷键,还具有转糖基化活性。fcn1基因的核苷酸序列如seq id no.1所示,其编码的氨基酸序列如seq id no.2所示。
[0039]
实施例2:fcn1基因克隆
[0040]
1、flavobacteriaceae bacterium rc2-3菌株基因组提取
[0041]
提取flavobacteriaceae bacterium rc2-3菌株基因组,取培养好的flavobacteriaceae bacterium rc2-3菌液,按照tiangen公司的细菌基因组dna提取试剂盒说明书使用试剂盒进行细菌基因组dna的提取,提取出的基因组dna用琼脂糖凝胶电泳进行测定。
[0042]
2、fcn1基因引物设计
[0043]
利用primer premier设计fcn1基因pcr引物,引物序列为:
[0044]
fcn1-f:5
’‑
ggtaccatgaataaactaatttcaatatttctaggaggg-3’(seq id no.3);
[0045]
fcn1-r:5
’‑
ggatccttaatctaaccaagtaattttagcttc-3’(seq id no.4)。
[0046]
3、q5超保真dna聚合酶pcr扩增
[0047]
使用mj mini个人型pcr仪,使用q5超保真dna聚合酶进行fcn1基因的扩增,反应体系如下:
[0048][0049][0050]
pcr扩增程序为:98℃预变性30s;94℃变性10s,66℃,退火15s,72℃延伸1min,30个循环;72℃延伸5min。
[0051]
pcr结束后以琼脂糖凝胶电泳方式回收fcn1基因。
[0052]
琼脂糖凝胶电泳验证结果如图3所示,fcn1基因扩增片段大小为1221bp。
[0053]
4、琼脂糖凝胶dna回收
[0054]
通过紫外光下切割包含目的dna片段的琼脂糖凝胶,按照tiangen公司的琼脂糖凝胶dna回收试剂盒说明书使用试剂盒进行目的dna片段的回收,回收后进行琼脂糖凝胶电泳验证并判断回收浓度。
[0055]
回收结果如图4所示,证明回收fcn1基因片段正确。
[0056]
5、fcn1-plb载体构建
[0057]
将plb与fcn1片段按照tiangen公司的plb零背景快速克隆试剂盒说明书,进行平末端连接,连接体系如下:
[0058][0059]
6、大肠杆菌转化
[0060]
(1)将含有100μg/ml氨苄霉素的lb固体平板,在37℃培养箱放置30min;
[0061]
(2)将10.0μl连接产物加入至100.0μl dh5α感受态细胞,轻柔混匀后冰浴静置
30min;
[0062]
(3)冰浴静置结束后42℃水浴热激90s,迅速转移至冰中静置1-3min;
[0063]
(4)加入400μl预热lb液体培养基,37℃,160rpm摇床中培养45min;
[0064]
(5)取上述菌液100μl涂布于预热的lb平板上,37℃倒置培养12-16h。
[0065]
利用plb零背景快速克隆试剂盒对dh5α菌株进行转化,培养12h后,在含有100μg/ml氨苄霉素抗性lb平板上生长出了重组菌株,而原始菌株dh5α由于缺少氨苄霉素抗性基因,不能在lb平板上生长,plb载体本身含有致死基因未连接fcn1基因的空质粒重组菌株无法生长,最终生长结果如图5所示。
[0066]
7、阳性转化子筛选
[0067]
挑取长出的菌落,接种于含100μg/ml氨苄霉素的lb液体培养基中,37℃,180rpm,培养6-8h。以plb-simple vector通用引物进行菌落pcr验证,pcr反应体系如下:
[0068][0069]
pcr扩增条件:94℃预变性5min;94℃变性30s,58℃,退火30s,72℃延伸2min,30个循环;72℃延伸5min。
[0070]
结果如图6所示,从图看出左边大部分菌落在1600bp和1000bp之间存在条带证明其中含有fcn1基因为阳性转化子,在1600bp与1000bp之间不存在条带的证明其中不含fcn1基因为假阳性。
[0071]
将得到的阳性转化子菌液保种后使用tiangen公司的质粒小提试剂盒提取质粒,将提取质粒送到青岛擎科生物技术有限公司进行测序,得到测序结果后与fcn1基因序列进行比对验证。质粒验证结果正确,其可以用于下一步实验。
[0072]
实施例3:fcn1基因异源表达及催化底物特异性初步探究
[0073]
1、fcn1基因异源表达载体构建
[0074]
选取实施例2测序完全正确的质粒,通过酶切、连接的方法将fcn1基因序列连接入表达载体pet-28a(+),完成rc2-3菌株fcn1基因表达载体fcn1-pet-28a(+)的构建。
[0075]
根据已经设计好的酶切位点,分别对pet-28a(+)载体和plb质粒进行双酶切,酶切体系如下:
[0076]
[0077][0078]
37℃条件下酶切2h,胶回收目的片段后,用t4 dna连接酶将二者连接,连接体系如下:
[0079][0080]
验证结果如图7,从图看出fcn1片段位于1600bp和1000bp之间证明可以切胶回收fcn1基因片段和pet-28a(+)载体。
[0081]
22℃水浴连接4h,连接后进行e.coli dh5α大肠杆菌转化,阳性转化子的筛选及质粒的提取,得到新质粒命名为fcn1-pet-28a(+)重组质粒,将新得到的fcn1-pet-28a(+)重组质粒转入到大肠杆菌菌株bl21中进行蛋白表达。
[0082]
2、大肠杆菌诱导表达
[0083]
(1)将验证获得的大肠杆菌重组菌株接种到5ml含有氨苄lb液体培养基中,37℃,200r/min培养过夜获得种子液;
[0084]
(2)将种子液按照1%的接种量转移到含有氨苄的50ml tb液体培养基中,37℃,200r/min培养至od600达到1.0-2.0;
[0085]
(3)添加终浓度为0.2%l-阿拉伯糖在16℃,200r/min培养12-16h以诱导蛋白表达;
[0086]
(4)4000r/min、4℃离心20min收集菌体,超声破碎菌体,12000r/min离心30min,取菌超声破碎后的上清进行sds-page检测,确定蛋白是否可溶性表达。
[0087]
3、sds-page凝胶电泳
[0088]
将样品制备:将蛋白质样品与5
×
样品缓冲液在一个eppendorf管中混合,放入100℃加热10min,取上清点样。
[0089]
分离胶及浓缩胶的制备,将玻璃板、样品梳、spacer用洗涤剂洗净,用ddh2o冲洗数次,再用乙醇擦拭,晾干。
[0090]
将两块洗净的玻璃板之间加入spacer,按照bio-rad mini li/ii说明书提示装好玻璃板。
[0091]
按照以下体系配置sds-page电泳所用凝胶:
[0092][0093]
首先向玻璃板间灌注分离胶,灌注结束后向立即覆盖一层无水乙醇,约20min后胶聚合,分离胶聚合后,将上层无水乙醇倾去,灌制浓缩胶,插入样品梳,等待胶凝固待胶完全凝固后,装好电泳系统,加入电极缓冲液,上样20μl;稳压200v,溴酚蓝刚跑出分离胶时,停止电泳,约需45min~60min。卸下胶板,剥离胶放入染色液中,室温染色1~2h;加入脱色液,置于80rpm脱色摇床上,每20min更换一次脱色液(10ml冰乙酸;45ml乙醇;45ml蒸馏水)至完全脱净。
[0094]
检测结果如图8所示,由fcn1基因指导编码的蛋白质预测大小为46.8kda,由图中观察可知在43kda上方1到4号组条带均比左一条带多出一个条带,证明fcn1基因在大肠杆菌bl21中实现了异源表达,并且fcn1基因指导合成的蛋白质在异源表达过程中没有形成包涵体,形成了有活性的蛋白。
[0095]
4、粗酶提取
[0096]
将发酵结束的发酵液分装至离心管中,于4℃,5000r/min条件下离心15min,取沉淀用适量预冷(4℃)蒸馏水搅拌清洗已得到相对洁净的菌体,在以相同的条件进行离心操作。重复上述步骤三次,最后取沉淀,得菌体。
[0097]
取菌体重的5倍体积的蒸馏水复溶,在冰浴条件下进行超声破碎,条件为时间15min,工作间歇为5s,结束后4℃,8000r/min离心取上清得到粗酶液,10ml/管分装在离心管中于-20℃下存放。
[0098]
5、酶学底物特异性分析
[0099]
配置多糖溶液,按照0.1006g多糖溶于50ml 1
×
pbs缓冲液的体系分别配置,岩藻多糖溶液、海带浓缩粉多糖溶液和岩藻聚糖硫酸酯多糖溶液。
[0100]
取2ml的粗酶液,在沸水浴中煮沸10min,将酶灭活,按照多糖溶液与灭活酶液/未灭活酶液1:1加体系配置反应体系,每种多糖溶液的灭活组与未灭活组各设置三个平行样,在设置条件为30℃、120r/min的培养箱中反应2h。
[0101]
取反应结束后溶液0.5ml加入铁氰化钾工作液0.5ml,在80℃水浴反应15min,室温冷却后加入2ml蒸馏水混匀,在10000pm离心10min,取上清液在420nm测定吸光度,空白为以蒸馏水代替样品测定的吸光度。
[0102]
表1:不同样品的吸光度
[0103][0104]
结果如表1所示,以岩藻多糖为底物和以海带浓缩粉为底物的灭活组与未灭活组od值差异不明显,说明fcn1基因编码的酶,对岩藻多糖和海带浓缩粉的降解能力不明显,无法产生大量还原糖与铁氰化钾发生反应导致,灭活酶组与未灭活酶组od值差距不明显。
[0105]
以岩藻聚糖硫酸酯为底物的实验组,未灭活组od值远大于灭活组od值,说明fcn1基因编码的酶对岩藻聚糖硫酸酯有着明显的降解能力,产生了大量还原糖与铁氰化钾发生反应使得未灭活组od值远大于灭活组od值。
[0106]
通过上述分析可知,fcn1基因编码的酶对岩藻多糖和海带浓缩粉降解能力不明显,对岩藻聚糖硫酸酯有着明显的降解能力。
[0107]
目前已知结构的岩藻聚糖硫酸酯的主链结构主要为
→
3-α-l-fuc-1
→
或是交替的
→
3-α-l-fuc-1
→
及
→
4-α-l-fuc-1
→
,而
→
2-α-l-fuc-1
→
有时作为支链出现,岩藻聚糖硫酸酯中主要以α1-3糖苷键为主,对应对fcn1的蛋白序列比对结果,fcn1蛋白序列与funa(wp_068826898.1)蛋白序列同源性达到70.98%并且氨基酸序列系统发育树显示两种酶亲缘关系最近来看,fcn1基因编码的蛋白有着降解α1-3糖苷键的能力。
[0108]
以上实施例仅用以说明本发明的技术方案,而非对其进行限制;尽管参照前述实施例对本发明进行了详细的说明,对于本领域的普通技术人员来说,依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或替换,并不使相应技术方案的本质脱离本发明所要求保护的技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1