一种恩赛特韦的合成方法与流程
1.本发明涉及化合物合成技术领域,具体涉及一种恩赛特韦的合成方法。
背景技术:
[0002][0003]
恩赛特韦即ensitrelvir是日本盐野义公司和北海道大学联合研发的针对新型冠状病毒的小分子药物。这是一种3cl蛋白酶抑制剂,主要用于抑制新冠病毒及各种变异毒株的活性,从而达到治疗新冠的目的。不同于辉瑞paxlovid (pf-07321332),恩赛特韦能够摆脱对p450酶抑制剂(如利托那韦)的依赖,实现单药治疗新冠,无需顾忌同时需服用的其他药物因p450酶抑制作用而产生的药理反应。
[0004]
其结构式为:
[0005][0006]
目前仅有biorxiv数据库中的文章《discovery of s-217622,a non-covalent oral sars-cov-2 3cl protease inhibitor clinical candidate for treating covid-19》报道了恩赛特韦的合成方法,合成路线如下:
[0007][0008]
文章中以硫脲和溴乙烷为原料,发生取代反应生产中间体a-1。a-1经关环和上叔丁基保护得到中间体a-2。a-2经过取代反应得到中间体a-3,再脱保护得到中间体a-4,最后经过两步取代反应分别得到中间体a-5和最终产物a-6(即恩赛特韦),总收率约5.1%。该路线较长,试剂价格昂贵,收率偏低,生产成本较高。原料气味难闻,使用三氟乙酸脱保护基对设备腐蚀性较强,不利于工业化生产。
技术实现要素:
[0009]
针对现有技术方案的不足,本发明旨在提供一种简化合成步骤,原料价格低,收率较高,适合工业化生产的恩赛特韦小分子药物的合成方法。
[0010]
为实现上述目的,本发明提供了一种恩赛特韦的合成方法,其合成方法为:
[0011]
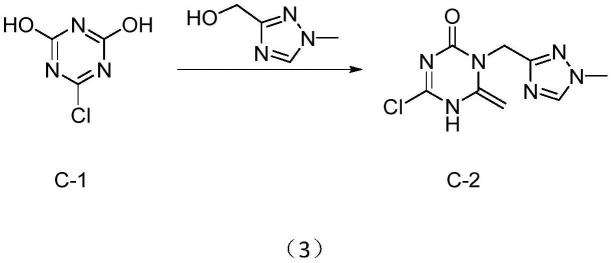
以三聚氯氰(sm)为原料水解反应得到中间体c-1;中间体c-1与(1-甲基
ꢀ‑
1h-1,2,4-三唑-3-基)甲醇反应得到中间体c-2;中间体c-2反应得到中间体c-3;中间体c-3反应得到产物c-4,即为恩赛特韦,反应方程式如下式(1)所示:
[0012][0013]
上述合成方法中所述的sm至c-1的水解反应在碱性或酸性环境下进行;所述的碱选自氢氧化钠、氢氧化钾、氢氧化锂中的一种或几种;所述的酸为盐酸、硫酸、磷酸、甲酸、乙酸中的一种或几种;优选为氢氧化钠或硫酸中的一种。
[0014]
所述的c-1至c-2的反应需在含有反应试剂的条件下进行,所述的反应试剂选自偶氮二甲酸二乙酯或偶氮二甲酸二异丙酯。
[0015]
优选地,所述的反应试剂为偶氮二甲酸二异丙酯。
[0016]
上述合成方法中所述的中间体c-2至c-3的反应是中间体c-2与2,4,5-三氟苄基溴在碱作用下反应;所述的碱选自碳酸钾、碳酸钠、碳酸铯、磷酸钾、甲醇钠、乙醇钠、叔丁醇钠、甲醇钾、乙醇钾、叔丁醇钾、三乙胺和吡啶中的一种或几种;优选为碳酸钾或甲醇钠。
[0017]
上述合成方法中所述的中间体c-3再反应得到产物c-4是中间体c-3和6
‑ꢀ
氯-2-甲基-2h-吲唑-5-胺在碱作用下反应;所述的碱选自碳酸钾、碳酸钠、磷酸钾、叔丁醇钾、叔丁醇钠、甲醇钠、乙醇钠、甲醇钾、乙醇钾、双三甲基硅基胺基锂、双三甲基硅基胺基钠和双三甲基硅基胺基钾中的一种或多种;优选为双三甲基硅基胺基锂或碳酸钾。
[0018]
一种恩赛特韦的合成方法,其合成方法具体包括如下步骤:
[0019]
(1)中间体c-1的制备:将三聚氯氰加入水或醇中加热反应,反应结束后得到中间体c-1,反应方程式如下式(2)所示:
[0020][0021]
(2)中间体c-2的制备:
[0022]
将步骤(1)中得到的中间体c-1、三苯基膦和(1-甲基-1h-1,2,4-三唑-3-基) 甲醇加入到溶剂1中,加入反应试剂,反应结束后加结晶溶剂得到中间体c-2,反应方程式如下式(3)所示:
[0023][0024]
(3)中间体c-3的制备:
[0025]
将步骤(2)中得到的中间体c-2与2,4,5-三氟苄基溴和碱加入到溶剂2中,加热条件下反应,反应结束加结晶溶剂得到中间体c-3,反应方程式如下式(4) 所示:
[0026][0027]
(4)恩赛特韦(中间体c-4)的制备:步骤(3)中得到的中间体c-3、6
‑ꢀ
氯-2-甲基-2h-吲唑-5-胺加入到溶剂3中,加入碱,反应结束后得到恩赛特韦即c-4,反应方程式如下式(5)所示:
[0028][0029]
步骤(1)中所述的加热反应在碱性或者酸性条件下进行,所述的碱为氢氧化钠、氢氧化钾和氢氧化锂的一种或几种;优选为氢氧化钠;所述的酸为盐酸、硫酸、磷酸、甲酸、乙酸中的一种或几种;优选为硫酸。
[0030]
步骤(2)中所述的反应试剂选用偶氮二甲酸二乙酯或偶氮二甲酸二异丙酯。
[0031]
优选地,步骤(2)中所述的反应试剂选用偶氮二甲酸二异丙酯。
[0032]
步骤(2)中所述的溶剂1为四氢呋喃,乙醚,乙酸乙酯,乙腈,n,n-二甲基甲酰胺,二氯甲烷和甲苯的一种或几种。
[0033]
步骤(2)和步骤(3)中所述的结晶溶剂为甲基叔丁基醚,乙醚,乙酸乙酯,正庚烷,石油醚,乙腈,二氯甲烷和甲苯的一种或几种。
[0034]
优选地,步骤(2)和步骤(3)中所述的结晶溶剂为乙酸乙酯、甲基叔丁基醚和甲苯中的一种或几种。
[0035]
步骤(3)中所述的碱为碳酸钾、碳酸钠、碳酸铯、磷酸钾、甲醇钠、乙醇钠、叔丁醇钠、甲醇钾、乙醇钾、叔丁醇钾、三乙胺和吡啶的一种或多种。
[0036]
优选地,步骤(3)中所述的碱为碳酸钾或/和甲醇钠。
[0037]
步骤(4)中所述的碱包括碳酸钾、碳酸钠、磷酸钾、叔丁醇钾、叔丁醇钠、甲醇钠、乙醇钠、甲醇钾、乙醇钾、双三甲基硅基胺基锂、双三甲基硅基胺基钠和双三甲基硅基胺基钾,的一种或多种。
[0038]
优选地,步骤(4)中所述的碱为双三甲基硅基胺基锂或/和碳酸钾。
[0039]
综上所述,本发明具有以下有益效果:
[0040]
第一、采用廉价易得的三聚氯氰(sm)为原料,避免合成原研路线中关三嗪环的繁琐步骤;
[0041]
第二、中间体c-1与(1-甲基-1h-1,2,4-三唑-3-基)甲醇的反应,选择性的与6 位上的氮反应,避免原研路线中采用叔丁基保护2位或者4位上的氮,再脱保护基的繁琐步骤。
[0042]
第三、操作简单、对设备腐蚀性较低,产物收率高适合工业化生产。
具体实施方式
[0043]
以下,根据本发明具体的实施形态进行说明。但是本发明不仅限定于该说明,在不脱离本发明的范围内,根据业者的知识,进行各种变更、修正及改良后而得的结果。
[0044]
实施例1中间体c-1的合成方法
[0045]
具体包括以下步骤:
[0046]
向1000g水中加入184g三聚氯氰,加热至40℃,搅拌下加入25%的氢氧化钠水溶液,直至溶液ph值维持在12-13,继续搅拌2小时。反应结束滴加盐酸调节ph值至6-7,溶液冷却至5-10℃,继续搅拌0.5小时,过滤,水淋洗,干燥得到130.9g中间体c-1为白色固体,hplc纯度99.3%,收率88.7%。
[0047]
实施例2中间体c-1的合成方法
[0048]
具体包括以下步骤:
[0049]
向1000g水中加入184g三聚氯氰,加热至50℃,搅拌下加入25%的稀硫酸,直至溶液ph值维持在1,继续搅拌1小时。反应结束滴加碳酸氢钠水溶液调节 ph值至6-7,溶液冷却至5-10℃,继续搅拌0.5小时,过滤,水淋洗,干燥得到 119.6g中间体c-1为白色固体,hplc纯度96.5%,收率81.1%。
[0050]
实施例3中间体c-1的合成方法
[0051]
具体包括以下步骤:
[0052]
向1000g水中加入184g三聚氯氰,加热至25℃,搅拌下加入25%的氢氧化锂水溶液,直至溶液ph值维持在12-13,继续搅拌15小时。反应结束滴加盐酸调节ph值至6-7,溶液
冷却至5-10℃,继续搅拌0.5小时,过滤,水淋洗,干燥得到115.5g中间体c-1为白色固体,hplc纯度97.7%,收率78.3%。
[0053]
1-h nmr(400mhz,dmso-d6)δ11.95(2h,br).
[0054]
实施例4中间体c-2的合成方法
[0055]
具体包括以下步骤:
[0056]
将实施例1所得的中间体c-1(29.6g,0.2mol)、三苯基膦(62.8g,0.24mol) 和(1-甲基-1h-1,2,4-三唑-3-基)甲醇(22.6g,0.2mol)加入500ml四氢呋喃中,氮气置换保护,冰水浴冷却,搅拌下缓慢滴加偶氮二甲酸二异丙酯(48.4g,0.24mol)。滴加完毕后升至室温,继续搅拌3小时,tlc显示原料反应完,反应液浓缩干,加600ml乙酸乙酯,200ml水洗有机相,无水硫酸钠干燥有机相,过滤,浓缩得到固体,加入200ml乙酸乙酯,搅拌1小时,过滤,少量乙酸乙酯淋洗,固体干燥得到35.6g中间体c-2,hplc纯度94.3%,收率73.3%。
[0057]
实施例5中间体c-2的合成方法
[0058]
具体包括以下步骤:
[0059]
将实施例1所得的中间体c-1(14.8g,0.1mol)、三苯基膦(31.4g,0.12mol) 和(1-甲基-1h-1,2,4-三唑-3-基)甲醇(11.3g,0.1mol)加入250ml乙醚中,氮气置换保护,冰水浴冷却,搅拌下缓慢滴加偶氮二甲酸二乙酯(20.9g,0.12mol)。滴加完毕后升至室温,继续搅拌3小时,tlc显示原料反应完,反应液浓缩干,加300ml乙酸乙酯,100ml水洗有机相,无水硫酸钠干燥有机相,过滤,浓缩得到固体,加入100ml甲基叔丁基醚,搅拌1小时,过滤,少量乙酸乙酯淋洗,固体干燥得到14.9g中间体c-2,hplc纯度92.7%,收率61.3%。
[0060]
1-h nmr(400mhz,dmso-d6)δ3.88(3h,s),5.09(2h,s),6.71(1h,s),10.66(1h, br).
[0061]
实施例6中间体c-3的合成方法
[0062]
具体包括以下步骤:
[0063]
将实施例4所得的中间体c-2(24.3g,0.1mol)、2,4,5-三氟苄基溴(23.6g, 1.05mol)和碳酸钾(20.7g,0.15mol)加入250ml甲苯中,升至室温85℃,搅拌3小时,tlc显示原料反应完。反应液加100ml甲苯稀释,150ml水洗有机相,无水硫酸钠干燥有机相,过滤,浓缩得到固体,加入50ml乙酸乙酯,搅拌 1小时,过滤,固体干燥得到36.7g中间体c-3,hplc纯度95.1%,收率94.8%。
[0064]
实施例7中间体c-3的合成方法
[0065]
具体包括以下步骤:
[0066]
将实施例4所得的中间体c-2(2.4g,10mmol)、2,4,5-三氟苄基溴(2.4g, 10.5mmol)和甲醇钠(4.9g,15mmol)加入50ml甲苯中,升至室温100℃,搅拌1小时,tlc显示原料反应完。反应液加50ml甲苯稀释,50ml水洗有机相,无水硫酸钠干燥有机相,过滤,浓缩得到固体,加入15ml甲苯,搅拌1小时,过滤,固体干燥得到3.4g中间体c-3,hplc纯度93.3%,收率88.3%。
[0067]
1-h nmr(400mhz,cdcl3)δ3.87(3h,s),5.13(2h,s),5.22(2h,s),6.67(1h,s), 6.91-6.97(1h,m),7.12-7.19(1h,m).
[0068]
实施例8恩赛特韦的合成方法
[0069]
具体包括以下步骤:
[0070]
将实施例7所得的中间体c-3(3.8g,0.01mol)和6-氯-2-甲基-2h-吲唑-5
‑ꢀ
胺(2.0g,0.011mol)加入50ml无水四氢呋喃中,氮气置换保护,冷却至0℃。搅拌下缓慢滴加1mol/l的双三甲基硅基胺基锂(15ml,0.015mol),约1小时滴加完毕,继续在0℃下搅拌1小时,升至室温搅拌1小时。反应液加入到60ml 饱和氯化铵溶液中淬灭,30ml乙酸乙酯萃取两次,合并有机相,无水硫酸钠干燥有机相,过滤,浓缩干,硅胶柱层析纯化(甲醇:二氯甲烷=5%~15%),得到4.7g恩赛特韦(c-4)固体,hplc纯度98.9%,收率88.1%。
[0071]
实施例9恩赛特韦的合成方法
[0072]
具体包括以下步骤:
[0073]
将实施例7所得的中间体c-3(3.8g,0.01mol)和6-氯-2-甲基-2h-吲唑-5
‑ꢀ
胺(2.0g,0.011mol)加入50ml无水四氢呋喃中,氮气置换保护,冷却至0℃。搅拌下缓慢滴加1mol/l的碳酸钾(15ml,0.015mol),约1小时滴加完毕,继续在0℃下搅拌1小时,升至室温搅拌1小时。反应液加入到60ml饱和氯化铵溶液中淬灭,30ml乙酸乙酯萃取两次,合并有机相,无水硫酸钠干燥有机相,过滤,浓缩干,硅胶柱层析纯化(甲醇:二氯甲烷=5%~15%),得到1.9g恩赛特韦(c-4)为淡棕色固体,hplc纯度98.7%,收率34.8%。
[0074]
实施例10恩赛特韦的合成方法
[0075]
具体包括以下步骤:
[0076]
将实施例7所得的中间体c-3(3.8g,0.01mol)和6-氯-2-甲基-2h-吲唑-5
‑ꢀ
胺(2.0g,0.011mol)加入50ml无水四氢呋喃中,氮气置换保护,冷却至0℃。搅拌下缓慢滴加1mol/l的双三甲基硅基胺基钾(15ml,0.015mol),约1小时滴加完毕,继续在0℃下搅拌1小时,升至室温搅拌1小时。反应液加入到60ml 饱和氯化铵溶液中淬灭,30ml乙酸乙酯萃取两次,合并有机相,无水硫酸钠干燥有机相,过滤,浓缩干,硅胶柱层析纯化(甲醇:二氯甲烷=5%~15%),得到3.0g恩赛特韦(c-4)固体,hplc纯度96.7%,收率55.8%。
[0077]
1-h nmr(400 mhz,pyridine-d5)δ3.89(3h,s),4.13(3h,s),5.06(2h,s),5.28 (2h,s),7.47(1h,m),7.53-7.67(2h,m),7.75(1h,s),8.43(1h,s),9.34(1h,s)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1