植物油基多元醇及其制备方法、热固化植物油基聚氨酯及其制备方法和应用与流程
1.本发明属于高分子材料技术领域,具体涉及一种植物油基多元醇及其制备方法、热固化植物油基聚氨酯及其制备方法和应用。
背景技术:
2.植物油是含有双键的长碳链的甘油三酯,其广泛分布于自然界中,种类繁多,其中常见的包括桐油、大豆油、亚麻油、玉米油、菜籽油、花生油、橄榄油、棕榈油、蓖麻油等。植物油因其特殊结构和可降解性,由其得到的聚合物不仅成本低,而且对环境友好,广泛应用于聚氨酯、生物柴油、涂料、生物医用等多个领域。
3.生物基多元醇可取代部分石油基多元醇用于制备聚氨酯,目前已研发的天然植物油基聚氨酯产品有粘结剂、油墨、涂料、润滑剂等。大豆油、棕榈油、棉籽油、葵花籽油等植物油都可以制备植物油基多元醇,再用作制备聚氨酯的原料。植物油基多元醇的使用能够降低对石油资源的依赖,同时产品可降解,对生态环境影响小。
4.现有技术中,公开号cn106957241b的专利公开一种高羟值桐油多元醇及其制备方法,其制备方法为:(1)将桐油、羧酸、酸催化剂、羟基化试剂、去离子水按比例混合,升温至35-45℃;在剧烈搅拌下滴加过氧化氢溶液,控制滴加速度使反应温度维持在40-65℃,滴加完毕后,维持反应温度2-8h;反应结束后静置分层,分离出水相,得到油相;(2)将步骤(1)的油相和含羟基胺类试剂按一定比例混合,加入乙醇、无机酸催化剂,在氮气保护下升温至30-40℃;在剧烈搅拌下滴加醛类试剂,滴加完毕后,在剧烈搅拌下升温至50-90℃,维持反应温度1-5h;反应结束后静置分层,油相经中和、水洗、减压蒸馏,得到高羟值桐油多元醇。虽然该方法制得的桐油多元醇具有较高的羟值,但是其制备方法较复杂,而且需要用到醛类试剂,醛类试剂无论是对人体还是对环境都有很大的不良影响。因此,有必要开发一种环境友好、制备工艺简单可控的植物油基多元醇的制备方法。
技术实现要素:
5.本发明的第一个目的在于提供一种植物油基多元醇的制备方法,本发明的第二个目的在于提供该制备方法制得的植物油基多元醇,本发明的第三个目的在于提供利用该植物油基多元醇制备热固化植物油基聚氨酯的制备方法,本发明的第四个目的在于提供该制备方法制得的热固化植物油基聚氨酯,本发明的第五个目的在于提供该热固化植物油基聚氨酯的应用。
6.根据本发明的第一个方面,提供了一种植物油基多元醇的制备方法,包括如下步骤:
7.将植物油、二乙醇胺、第一催化剂在70-80℃下反应2-6小时,当反应产物的酸值低于5mg koh/g时,可判定此步反应达到终点,得到胺解植物油;
8.将胺解植物油、酸酐、第二催化剂在110-210℃下反应3-6小时,当反应产物的酸值
低于30mg koh/g时,可判定此步反应达到终点,然后降温至70-100℃,加入环氧植物油,混合均匀,即得。
9.植物油的酯基可以发生胺解反应,最终得到的产物是酰胺,植物油胺解反应的示意图如图1所示,图1中,最左端的反应物代表植物油,箭头上的反应物代表胺解剂。植物油胺解反应得到的酰胺可以通过后续聚合、环氧化等反应赋予材料不同的力学性能。
10.本发明先利用二乙醇胺对植物油进行胺解制备胺解植物油,以在植物油上引入多个羟基,提高反应活性位点,因而胺解植物油具备与酸酐进行开环反应的基础;然后利用酸酐对胺解植物油进行酯化改性,并引入六元环刚性结构,以提高由植物油基多元醇制得的聚氨酯的力学性能;最后加入环氧植物油进行共混,目的在于降低植物油基多元醇的粘度,增加由植物油基多元醇制得的聚氨酯的韧性。
11.由于对比三乙醇胺,二乙醇胺的反应活性更高;对比n-甲基乙醇胺,二乙醇胺具备双羟基,能在反应产物中引入更多羟基,有利于后续产品的性能优化;而且二乙醇胺价格低廉、活性较高、简单易得,因此,本发明选择二乙醇胺作为胺解剂。
12.当植物油、二乙醇胺胺解反应的温度为70-80℃时,胺解反应的反应速率较快且副产物少;当温度高于80℃时,胺解反应的反应速率没有得到提高而且还会生成大量副产物。因此,本发明将植物油、二乙醇胺的反应温度设置为70-80℃。
13.在一些实施方式中,胺解植物油和酸酐反应的升温速率为每半个小时10℃,升温至150℃时停止升温,测定反应产物的酸值。若酸值≤30mg koh/g,可判定此步反应达到终点;若酸值>30mg koh/g,继续升温至210℃时停止升温,测定反应产物的酸值。
14.在一些实施方式中,当反应体系的酸值持续不变时,可以向反应体系中添加适量的催化剂。
15.在一些实施方式中,植物油中的酯键与二乙醇胺的用量比例为1mol:(0.8-2.0)mol,胺解植物油中的羟基与酸酐的用量比例为1mol:(1.0-2.5)mol,环氧植物油的用量为原料总质量的10%-50%。
16.在一些实施方式中,植物油中的酯键与二乙醇胺的用量比例为1mol:(1.0-1.2)mol,胺解植物油中的羟基与酸酐的用量比例为1mol:(1.0-1.5)mol,环氧植物油的用量为原料总质量的20%-30%。
17.在一些实施方式中,第一催化剂为钛酸正丁酯(tbt)、氢氧化钠(naoh)、氢氧化钾(koh)中的任意一种或一种以上任意比例的混合物,第一催化剂的用量为原料总质量的1.0-3.6%。
18.在一些实施方式中,第二催化剂为苄基三乙基氯化铵、钛酸正丁酯、三乙胺、三苯基膦中的任意一种或一种以上任意比例的混合物,第二催化剂的用量为原料总质量的1.0-3.6%。由此,可以避免胺解植物油和酸酐反应产生的羧基使氨基质子化从而影响反应的进行。
19.在一些实施方式中,植物油为大豆油、蓖麻油、桐油、亚麻油、菜籽油中的任意一种或一种以上任意比例的混合物;酸酐为苯酐、卤代苯酐、四氢苯酐、甲基四氢苯酐、甲基六氢苯酐中的任意一种或一种以上任意比例的混合物;环氧植物油为环氧大豆油、环氧亚麻油、环氧蓖麻油中的任意一种或一种以上任意比例的混合物。
20.本发明选择带六元环结构的酸酐,由此,在植物油基多元醇结构中引入六元环,后
续利用植物油基多元醇制备聚氨酯时,六元环能赋予聚氨酯刚性结构,提高聚氨酯的力学性能。
21.本发明加入环氧植物油的目的是降低反应产物的粘度,同时增加由植物油基多元醇制备的聚氨酯的韧性。
22.根据本发明的第二个方面,提供了上述的植物油基多元醇的制备方法制得的植物油基多元醇。
23.根据本发明的第三个方面,提供了一种热固化植物油基聚氨酯的制备方法,包括如下步骤:
24.将上述的植物油基多元醇与异氰酸酯、第三催化剂搅拌混匀后,在60-70℃下干燥3-5h,即得;植物油基多元醇中的羟基与异氰酸酯的用量比例为1mol:(0.5-1.5)mol。
25.在一些实施方式中,植物油基多元醇中的羟基与异氰酸酯的用量比例为1mol:(0.8-1.2)mol。
26.在一些实施方式中,异氰酸酯为甲苯二异氰酸酯、异佛尔酮二异氰酸酯、二苯基甲烷二异氰酸酯、二环己基甲烷二异氰酸酯、六亚甲基二异氰酸酯、赖氨酸二异氰酸酯中的任意一种或一种以上任意比例的混合物。由此,异氰酸酯的选择会影响后续产品的粘度、硬度、韧性、热稳定性等性能。
27.在一些实施方式中,第三催化剂为二月桂酸二丁基锡、三亚乙基二胺、辛酸亚锡中的任意一种或一种以上任意比例的混合物,第三催化剂的用量为原料总质量的0.05-1.0%。
28.在一些实施方式中,将植物油基多元醇与异氰酸酯、第三催化剂搅拌混匀后,用热鼓风干燥箱或烘箱进行干燥,干燥的温度为60℃,时间为3h。
29.根据本发明的第四个方面,提供了上述的热固化植物油基聚氨酯的制备方法制得的热固化植物油基聚氨酯。
30.根据本发明的第五个方面,提供了上述的热固化植物油基聚氨酯在制备油墨、粘结剂、地坪涂料中的应用。
31.本发明的有益效果包括:
32.(1)本发明首先通过胺解反应在植物油上引入羟基,提高反应活性位点,然后使胺解植物油与酸酐通过开环反应进行结合,再加入环氧植物油进行共混,得到植物油基多元醇。本发明得到的植物油基多元醇具有六元环刚性结构、带有端羟基,具有多个活性官能团可参与固化反应,如羟基与异氰酸酯、羧基与环氧基、羟基与羧基、羧基与异氰酸酯基等交联固化,因此具备与异氰酸基团反应固化的基础。将植物油基多元醇与异氰酸酯进行反应固化,制得的聚氨酯韧性增强的同时保持较高的硬度,且随着环氧植物油用量的不同,可对聚氨酯热固化膜的韧性进行规律性调控。
33.(2)本发明选择带六元环结构的酸酐对胺解植物油进行酯化改性,由此,在植物油基多元醇结构中引入六元环,后续利用植物油基多元醇制备聚氨酯时,六元环能赋予聚氨酯刚性结构,提高聚氨酯的力学性能。而且本发明的胺解植物油与酸酐的反应无需溶剂即可进行,省去了后续除去溶剂的步骤,降低了生产成本。
34.(3)本发明的植物油基多元醇中引入了大量的羟基、环状结构以及环氧植物油,可有效提高由植物油基多元醇制得的聚氨酯热固化膜的拉伸强度与韧性,热固化膜的储存稳
定性良好,交联密度高。
35.(4)本发明的热固化植物油基聚氨酯树脂的外观均匀不分层,稳定性良好,硬度较强,耐酸碱性良好,凝胶率良好,拉伸强度优秀,而且生物基含量高、生物降解性好、原料利用率高、生产工艺连续可控,适用于制备油墨、粘结剂、地坪涂料。
附图说明
36.图1为植物油胺解反应的示意图。
37.图2为本发明实施例1的步骤(1)的反应路线图。
38.图3为本发明实施例1的步骤(2)的反应路线图。
39.图4为本发明实施例1的蓖麻油、蓖麻油基多元醇的傅里叶变换红外谱图。
40.图5为本发明实施例8的反应路线图。
具体实施方式
41.下面结合具体实施例对本发明作进一步详细的说明,但本发明的实施方式不限于此。下列实施例中涉及的物料均可从商业渠道获得。
42.实施例1
43.本实施例的蓖麻油基多元醇的制备过程如下:
44.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺(dea)、6.8g催化剂氢氧化钾,机械搅拌,升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油(co)(蓖麻油中的酯键与二乙醇胺的用量摩尔比为1:1),在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油(codea)。该步骤的反应路线图如图2所示。
45.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油(codea),144g甲基四氢苯酐(mthpa)(胺解蓖麻油中所含羟基与甲基四氢苯酐的用量摩尔比为3:1),1g催化剂三苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入86g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇(codea-mthpa)。该步骤的反应路线图如图3所示。
46.本实施例的蓖麻油、蓖麻油基多元醇的傅里叶变换红外谱图如图4所示。从图4对比可以看到,原料为蓖麻油(记为co),产物为蓖麻油基多元醇(记为codea-mthpa),具体地:
47.在co的红外图可看出,其特征峰为3500cm-1
处长宽型吸收峰,为-oh伸缩振动。
48.在codea-mthpa的红外图可看出:3500cm-1
处-oh峰减弱,说明体系中-oh被消耗,-oh消耗越多说明反应越充分;1750cm-1
处-coo-峰减弱,说明体系中酯键断裂;1650cm-1
处出现-nhco-振动吸收峰,说明成功引入甲基四氢苯酐。因此,ft-ir结果表明,产物为蓖麻油基多元醇。
49.本实施例制得的蓖麻油基多元醇的酸值为10mg koh/g,羟值为125mg koh/g。
50.实施例2
51.本实施例的蓖麻油基多元醇的制备过程如下:
52.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺、6.8g催化剂氢氧化钾,机械搅拌,
升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油,在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油。
53.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油,144g甲基四氢苯酐,1g催化剂三苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入43g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇。
54.本实施例制得的蓖麻油基多元醇的酸值为22mg koh/g,羟值为73mg koh/g。
55.实施例3
56.本实施例的蓖麻油基多元醇的制备过程如下:
57.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺、6.8g催化剂氢氧化钾,机械搅拌,升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油,在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油。
58.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油,144g甲基四氢苯酐,1g催化剂三苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入64.5g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇。
59.本实施例制得的蓖麻油基多元醇的酸值为17mg koh/g,羟值为95mg koh/g。
60.实施例4
61.本实施例的蓖麻油基多元醇的制备过程如下:
62.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺、6.8g催化剂氢氧化钾,机械搅拌,升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油,在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油。
63.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油,144g四氢苯酐,1g催化剂三苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入86g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇。
64.本实施例制得的蓖麻油基多元醇的酸值为11mg koh/g,羟值为135mg koh/g。
65.实施例5
66.本实施例的蓖麻油基多元醇的制备过程如下:
67.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺、6.8g催化剂氢氧化钾,机械搅拌,升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油,在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油。
68.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油,144g四氢苯酐,1g催化剂三
苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入43g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇。
69.本实施例制得的蓖麻油基多元醇的酸值为26mg koh/g,羟值为70mg koh/g。
70.实施例6
71.本实施例的蓖麻油基多元醇的制备过程如下:
72.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺、6.8g催化剂氢氧化钾,机械搅拌,升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油,在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油。
73.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油,144g四氢苯酐,1g催化剂三苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入64.5g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇。
74.本实施例制得的蓖麻油基多元醇的酸值为22mg koh/g,羟值为85mg koh/g。
75.实施例7
76.本实施例的蓖麻油基多元醇的制备过程如下:
77.(1)在1000ml圆底烧瓶中投入152.5g二乙醇胺、6.8g催化剂氢氧化钾,机械搅拌,升温至70℃直至催化剂溶解。然后缓慢加入466.5g蓖麻油,在0.5h内加入完毕,然后保持温度不变,反应2小时,每隔0.5小时测定反应物酸值,当酸值小于5mg koh/g即达到反应终点。然后往圆底烧瓶中加入300g乙酸乙酯,随后将溶液转移至分液漏斗,用饱和食盐水洗涤3次,将有机相于45℃旋蒸除去溶剂,得到浅黄透明的胺解蓖麻油。
78.(2)在另一个1000ml圆底烧瓶中加入200g胺解蓖麻油,144g四氢苯酐,1g催化剂三苯基膦,升温至150℃,持续反应,每隔半小时测定一次酸值,直至酸值小于30mg koh/g。然后冷却至80℃,加入129g环氧大豆油,混合均匀,得到浅棕黄色透明的蓖麻油基多元醇。
79.本实施例制得的蓖麻油基多元醇的酸值为5mg koh/g,羟值为165mg koh/g。
80.下面,利用实施例1-7制得的蓖麻油基多元醇制备热固化蓖麻油基聚氨酯。需要说明的是,环氧大豆油的投料量会影响所得蓖麻油基多元醇的羟值,因此实施例1-7制得的蓖麻油基多元醇的羟值不同,且随环氧大豆油用量的变化而呈现规律性变化。利用多元醇制备聚氨酯时,异氰酸酯的投料量是根据多元醇的羟值来确定的,因此实施例8-14中,在蓖麻油基多元醇的投料量相同时,异氰酸酯的用量不同。
81.实施例8
82.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
83.将10g实施例1制得的蓖麻油基多元醇(codea-mthpa)与2.5g异佛尔酮二异氰酸酯(ipdi)在0.014g催化剂二月桂酸二丁基锡(dbtdl)作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。该步骤的反应路线图如图5所示。
84.实施例9
85.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
86.将10g实施例2制得的蓖麻油基多元醇与1.5g异佛尔酮二异氰酸酯在0.014g催化
剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
87.实施例10
88.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
89.将10g实施例3制得的蓖麻油基多元醇与1.9g异佛尔酮二异氰酸酯在0.014g催化剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
90.实施例11
91.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
92.将10g实施例4制得的蓖麻油基多元醇与2.7g异佛尔酮二异氰酸酯在0.014g催化剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
93.实施例12
94.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
95.将10g实施例5制得的蓖麻油基多元醇与1.4g异佛尔酮二异氰酸酯在0.014g催化剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
96.实施例13
97.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
98.将10g实施例6制得的蓖麻油基多元醇与1.7g异佛尔酮二异氰酸酯在0.014g催化剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
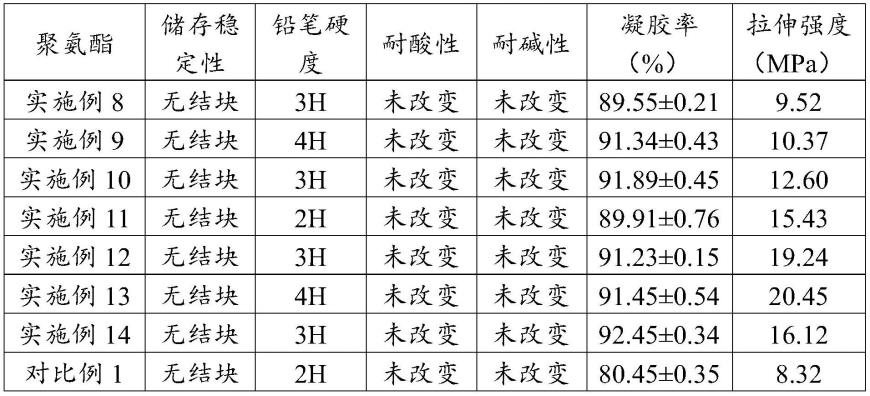
99.实施例14
100.本实施例的热固化蓖麻油基聚氨酯的制备过程如下:
101.将10g实施例7制得的蓖麻油基多元醇与3.3g异佛尔酮二异氰酸酯在0.014g催化剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
102.对比例1
103.本对比例的热固化蓖麻油基聚氨酯的制备过程如下:
104.将10g市售蓖麻油基改性多元醇(产品品牌:欧宝迪,产品型号:albodur 901)与3.7g异佛尔酮二异氰酸酯在0.014g催化剂二月桂酸二丁基锡作用下搅拌共混,混合均匀后放入热鼓风干燥箱中在60℃下干燥3h,得到热固化蓖麻油基聚氨酯。
105.为验证本发明的聚氨酯树脂的综合性能,下面对实施例8-14和对比例1制备得到的热固化蓖麻油基聚氨酯进行机械性能测试、储存稳定性测试、铅笔硬度测试、耐酸碱性测试和凝胶率测试。
106.1、测试方法
107.机械性能测试方法:采用深圳三思纵横通用测试机对样品进行分析,选用拉伸支架,样品的尺寸为40.0mm(长)
×
10.0mm(宽)
×
0.5mm(厚),十字头速度为10.00mm/min。为了准确起见,每种样品都进行三次测量并取平均值。
108.储存稳定性测试方法:将样品置于80℃鼓风烘箱中,观察有无结块。为了准确起见,每种样品都进行三次测量并取平均值。
109.铅笔硬度测试方法:用国标gb/t6739-1996法(硬度等级范围为6b~hb~6h,其中6h为最硬,6b为最软)测定样品的铅笔硬度。具体操作:硬度计使用三点接触法测定样品表面(两点为滚轮,一点为铅笔芯),铅笔与样品表面夹角为45
°
,使用硬度计在样品表面用压力为1
±
0.05kg的力滑行,观察样品的破损情况,当5次试验中不多于2次破损时,更换大一等级硬度的铅笔进行测试,当样品的破损超过2次时,则可读取此时铅笔等级并记录此等级的下一位等级。为了准确起见,每种样品都进行三次测量并取平均值。
110.耐酸碱性测试方法:精确称量样品0.100~0.300g,在室温下分别浸没在10%naoh水溶液、10%hcl水溶液中24h,然后取出样品观察溶解情况,并用吸水纸干燥样品后称重,观察样品质量是否改变。为了准确起见,每种样品都进行三次测量并取平均值。
111.凝胶率测试方法:在室温下,称取适量样品浸没于装有丙酮的密封玻璃瓶中,浸泡24小时后,将样品取出并置于60℃真空干燥箱中干燥至恒重,记录样品浸泡前的质量w0和干燥后的质量w1。凝胶率按照以下公式计算:凝胶率=w1/w0×
100%。为了准确起见,每种样品都进行三次测量并取平均值。
112.2、测试结果
113.实施例8-14及对比例1制备的聚氨酯的综合性能测试结果如表1所示:
114.表1实施例8-14及对比例1制备的聚氨酯的综合性能测试结果
[0115][0116]
从表1可以看出,本发明制备的热固化蓖麻油基聚氨酯的稳定性良好,在80℃下没有结块;铅笔硬度等级为2h~4h,硬度较强;在10%naoh水溶液或10%hcl水溶液中浸泡24h后质量未发生改变,说明耐酸碱性良好;凝胶率为89~92%,凝胶率良好,说明本发明制得的蓖麻油基聚氨酯的交联密度很好;拉伸强度可达到9~20mpa,对比对比例1的同类型高生物基含量聚氨酯,本发明制备的热固化蓖麻油基聚氨酯的拉伸强度优秀。
[0117]
从表1还可以看出,在实施例13的聚氨酯达到最大拉伸强度后,环氧大豆油添加量的增多会降低聚氨酯的拉伸强度,原因在于蓖麻油基多元醇中的羧基与环氧基反应完全后,环氧大豆油不再接入聚氨酯,环氧大豆油仅表现生物基增韧剂作用。因此,加入过多的环氧大豆油有利于提高聚氨酯的韧性但相对应会降低聚氨酯的拉伸强度。由此启示,利用本发明的植物油基多元醇制备聚氨酯时,可通过调节环氧植物油的添加量来调节聚氨酯的
刚性以使制备的聚氨酯适应不同的使用环境,由此可以扩宽聚氨酯的应用范围。
[0118]
以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1