缩合法生产光引发剂UV-1173的方法与流程
缩合法生产光引发剂uv-1173的方法
技术领域
1.本发明属于精细化工技术领域,特别涉及一种缩合法生产光引发剂uv-1173的方法。
背景技术:
2.光引发剂uv-1173(2-羟基-2-甲基-1-苯基-1-丙酮)是一种性能优良的新型光引发剂,具有引发效率高、热稳定性好、耐黄变、无异味等优点,在光固化中的地位尤为突出
3.以异丁酸为原料,依次经酰化反应、傅克反应、氯化反应、碱解反应制备2-羟基-2-甲基-1-苯基-1-丙酮,是光引发剂uv-1173的主要生产方法。例如,专利号为201110200666.8和201810893357.5的中国发明专利均记载了以类似工艺生产光引发剂uv-1173的过程。
4.然而,上述工艺不仅需要用到大量的危险化学品,如三氯化磷、三氯化铝及氯气等,且过程中产生大量三废物质,如高磷废水、由傅克反应水解处理产生的铝水、氯化氢气体及盐酸废水等,不利于环境友好。
5.为克服上述问题,专利号为201810727673.5的中国发明专利提供了一种光引发剂1173的制备工艺,以苯甲酸和异丁酸为原料,在金属盐催化剂作用下,高温脱水脱二氧化碳制得异丁酰苯,然后以所述异丁酰苯为中间产品、以四氯化碳和氢氧化钠为试剂、以四丁基溴化铵为相转移催化剂,进行一锅法氯化和碱解反应制得2-羟基-2-甲基-1-苯基-1-丙酮。上述工艺过程虽然减少了危险化学物质的使用,降低了三废的产生量,然而,受其催化剂及反应温度的选择影响,反应收率较低,且需要频繁更换催化剂,不仅不利于长时间稳定生产,且产生较大量固体废弃物。
技术实现要素:
6.基于此,本发明提供一种缩合法生产光引发剂uv-1173的方法,以解决现有技术中存在的以苯甲酸和异丁酸为原料生产光引发剂uv-1173时,收率较低,且不能长时间稳定生产的技术问题。
7.本发明解决上述技术问题的技术方案如下:
8.一种缩合法生产光引发剂uv-1173的方法,包括以下步骤:
9.s10.将异丁酸与苯甲酸混合,配置原料混合液a,其中,异丁酸与苯甲酸的物质的量的比为1:(0.6-1.0);
10.s20.原料混合液a与催化剂接触,在第一反应温度下,发生缩合反应,生成异丁酰苯;其中,催化剂为mn
2+
盐中的一种或多种;
11.s30.以异丁酰苯合成2-羟基-2-甲基-1-苯基-1-丙酮。
12.优选地,步骤s20中,所述“原料混合液a与催化剂接触,在第一反应温度下,发生缩合反应,生成异丁酰苯”包括以下步骤:
13.s21.催化剂活化阶段:将原料混合液a升温至活化温度,加入催化剂,并充分混合,
保温第一时长;
14.s22.缩合反应阶段:保温结束后,在第一反应温度下,滴加原料混合液a,进行缩合反应;冷凝收集气相馏分,得到粗品b;
15.s23.分离阶段:从粗品b中分离未反应的原料异丁酸与苯甲酸以及异丁酰苯。
16.优选地,所述第一反应温度为200℃-400℃。
17.优选地,步骤s21中,所述活化温度为200℃-400℃。
18.优选地,步骤s22中,保温结束后,在第一反应温度下,按每千克催化剂40g/h-75g/h的滴加速率,滴加原料混合液a,进行缩合反应。
19.优选地,步骤s21中,催化剂和原料混合液a的质量比1:(1-1.5)。
20.优选地,步骤s10中,异丁酸与苯甲酸的物质的量的比为1:(0.83-0.90)。
21.优选地,催化剂为mno、mncl、mn(no3)2、mnso4、mnco3、苯甲酸锰(ii)、乙酸锰(ii)的一种或多种。
22.优选地,步骤s30中,所述“以异丁酰苯合成2-羟基-2-甲基-1-苯基-1-丙酮”包括以下步骤:以异丁酰苯为原料,经过氯化反应和碱解反应过程,制备2-羟基-2-甲基-1-苯基-1-丙酮。
23.优选地,步骤s30中,所述“以异丁酰苯合成2-羟基-2-甲基-1-苯基-1-丙酮”包括以下步骤:
24.s31.氯化反应:在50℃-80℃的氯化反应温度下,向异丁酰苯中通入cl2,进行氯化反应,得到氯酮a;
25.s32.碱解反应:将氯酮a投加到液碱中,搅拌进行碱解反应。
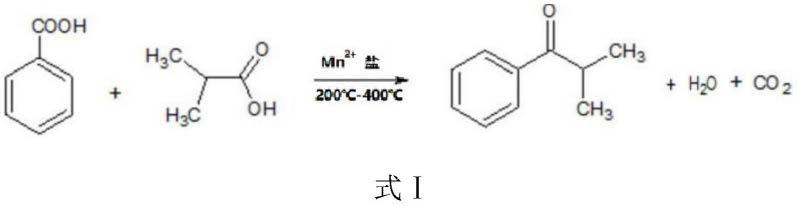
26.与现有技术相比,本发明至少具有以下优点:
27.以苯甲酸与异丁酸为原料,在mn
2+
盐存在下,于200℃-400℃的温度下发生缩合反应,一步生成中间产物异丁酰苯,然后对异丁酰苯进行氯化、碱解,制备2-羟基-2-甲基-1-苯基-1-丙酮(即光引发剂uv-1173)。本发明提供的方法一步法合成了异丁酰苯,相比傅克反应过程,减少了危险化学品的使用,降低了三废的产出量,是一种环境友好的生产工艺。以mn
2+
盐作为催化剂,实验表明,有利于提高光引发剂uv-1173的产率,uv-1173的收率可以达到90%以上(按苯甲酸计算)。同时,以mn
2+
盐作为催化剂,实现连续生产,有利于延长生产周期,实验表明,2.5kg的mn
2+
盐,连续投料200kg,未见催化剂失活。工业生产实践表明,2000kg的mn
2+
盐,连续投料400天,可生产异丁酰苯632t。催化剂失活后,产生约4200kg固体废弃物,即生产每吨异丁酰苯仅需要产生6.6kg固体废弃物,大幅度降低了固体废弃物的产生量。
具体实施方式
28.需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。以下将结合本发明实施例,对本发明的技术方案做进一步描述,本发明不仅限于以下具体实施方式。
29.一实施例中,一种缩合法生产光引发剂uv-1173的方法,包括以下步骤:
30.s10.将异丁酸与苯甲酸混合,配置原料混合液a,其中,异丁酸与苯甲酸的物质的量的比为1:(0.6-1.0)。
31.作为优选,异丁酸与苯甲酸的物质的量的比为1:(0.90-0.98),对应的苯甲酸与异丁酸的质量比为(1.25-1.35):1,也就是说,反应的原料体系中,异丁酸过量。
32.s20.原料混合液a与催化剂接触,在第一反应温度下,发生缩合反应,生成异丁酰苯;其中,催化剂为mn
2+
盐中的一种或多种。
33.作为优选,所述第一反应温度为200℃-400℃,进一步地,所述第一反应温度为280℃-320℃。缩合反应过程如式ⅰ所示:
[0034][0035]
在200℃-400℃温度下,以mn
2+
盐作为催化剂,苯甲酸和异丁酸缩合反应,生成异丁酰苯,并同时生成水和二氧化碳。在高温下,水和二氧化碳以气相形式被排出反应体系,有利于反应的进行。
[0036]
例如,催化剂mn
2+
盐可以是mno、mncl、mn(no3)2、mnso4、mnco3、苯甲酸锰(ii)、乙酸锰(ii)的一种或多种。例如,催化剂mn
2+
盐可以是mno和mnco3的混合物。作为优选,mn
2+
盐可以是苯甲酸锰(ii)。
[0037]
具体地,上述过程包括以下步骤:
[0038]
s21.催化剂活化阶段:将原料混合液a升温至活化温度,加入催化剂,并充分混合,保温第一时长。
[0039]
一具体实施方式中,首先将原料混合物a升温值活化温度,例如所述活化温度为200℃-400℃,冷凝并收集气相,建立全回流。升温至目标温度后,加入催化剂mn
2+
盐,作为优选,催化剂的加入量为原料混合物a质量的1-1.5倍。充分搅拌,使得催化剂mn
2+
盐与原料混合物a充分混合,保温6h-24h。
[0040]
s22.缩合反应阶段:保温结束后,在第一反应温度下,滴加原料混合液a,进行缩合反应;冷凝收集气相馏分,得到粗品b。
[0041]
向催化剂mn
2+
盐保温活化结束后的反应体系中滴加原料混合液a,进行缩合反应。冷凝收集气相馏分,部分被作为回流液相返回反应体系,部分被采出,作为含异丁酰苯的粗品。作为优选,所述第一反应温度200℃-400℃。原料混合液a应被缓慢滴加进入反应体系内,作为优选,按每千克催化剂40g/h-75g/h的滴加速率,滴加原料混合液a,进行缩合反应。值得说明的是,所述“按每千克催化剂40g/h-75g/h的滴加速率,滴加原料混合液a”应该被理解为:如果体系内的催化剂含量为1kg,则原料混合液a的滴加速率为40g/h-75g/h;如果体系内的催化剂含量为2kg,则原料混合液a的滴加速率为80g/h-150g/h;如果体系内的催化剂含量为1000kg,则原料混合液a的滴加速率为40kg/h-75kg/h,以此类推。
[0042]
s23.分离阶段:从粗品b中分离未反应的原料异丁酸与苯甲酸以及异丁酰苯。
[0043]
例如,采用蒸馏方法,从粗品b中分离原料异丁酸和中间产品异丁酰苯,其中,异丁酰苯的纯度≥99%。
[0044]
s30.以异丁酰苯合成2-羟基-2-甲基-1-苯基-1-丙酮。
[0045]
可以通过多种方式,以异丁酰苯为原料,合成2-羟基-2-甲基-1-苯基-1-丙酮。例如,可通过如专利号为201810727673.5的中国发明专利中提出的以异丁酰苯为原料以四氯化碳和氢氧化钠为试剂,在四丁基溴化铵为相转移催化剂的体系中,进行一锅法氯化和碱解反应制得2-羟基-2-甲基-1-苯基-1-丙酮。也可以采用如专利号201810893357.5的中国发明专利中记载的先氯化反应,得到氯酮,然后经过碱解反应,制备2-羟基-2-甲基-1-苯基-1-丙酮粗品。
[0046]
作为优选,步骤s30中,所述“以异丁酰苯合成2-羟基-2-甲基-1-苯基-1-丙酮”包括以下步骤:
[0047]
s31.氯化反应:在50℃-80℃的氯化反应温度下,向异丁酰苯中通入cl2,进行氯化反应,得到氯酮a。
[0048]
向异丁酰苯中通入cl2,进行氯化反应制备氯酮a(化学式如式ⅱ)。氯化过程中,产生的含hcl和cl2的尾气经水吸收后,副产盐酸。
[0049][0050]
s32.碱解反应:将氯酮a投加到液碱中,搅拌进行碱解反应。
[0051]
将氯酮a投加到液碱(naoh溶液,0.1m)中,搅拌进行碱解反应,当体系内的氯酮a含量小于0.2%时,碱解反应结束。加水洗涤至中性,得到2-羟基-2-甲基苯基丙烷-1-酮粗品。
[0052]
2-羟基-2-甲基苯基丙烷-1-酮粗品经精馏等进一步处理,得到纯度≥95%的2-羟基-2-甲基苯基丙烷-1-酮成品,即光引发剂uv-1173。
[0053]
值得说明的是,本领域技术人员应该可以理解的是,本发明的技术构思还可以被应用于制备其他α-羟基酮类光引发剂,如制备uv-184(1-羟基环己基苯基甲酮)、uv-2959(2-羟基-4'-(2-羟乙氧基)-2-甲基苯丙酮)、光引发剂185(2-羟基-2-甲基-1-[4-(叔丁基)苯基]-1-丙酮)。例如,以mn
2+
盐作为催化剂,以苯甲酸和环己甲酸为原料,用于制备1-羟基环己基苯基甲酮,进而制备光引发剂uv-184。
[0054]
再例如,一种光引发剂185的生产方法,包括以下步骤:
[0055]
t10.将异丁酸与叔丁基苯甲酸混合,配置原料混合液a,其中,异丁酸与叔丁基苯甲酸的物质的量的比为1:(0.6-1.0)。
[0056]
作为优选,异丁酸与叔丁基苯甲酸的物质的量的比为1:(0.83-0.90),也就是说,反应的原料体系中,异丁酸过量。
[0057]
t20.原料混合液a与催化剂接触,在第一反应温度下,发生缩合反应,生成2-甲基-1-[4-(叔丁基)苯基]-1-丙酮;其中,催化剂为mn
2+
盐中的一种或多种。
[0058]
作为优选,所述第一反应温度为200℃-400℃,进一步地,所述第一反应温度为280℃-320℃。
[0059]
在200℃-400℃温度下,以mn
2+
盐作为催化剂,叔丁基苯甲酸和异丁酸缩合反应,生成2-甲基-1-[4-(叔丁基)苯基]-1-丙酮,并同时生成水和二氧化碳。在高温下,水和二氧化
碳以气相形式被排出反应体系,有利于反应的进行。
[0060]
例如,催化剂mn
2+
盐可以是mno、mncl、mn(no3)2、mnso4、mnco3、苯甲酸锰(ii)、乙酸锰(ii)的一种或多种。例如,催化剂mn
2+
盐可以是mno和mnco3的混合物。作为优选,mn
2+
盐可以是苯甲酸锰(ii)。
[0061]
具体地,上述过程包括以下步骤:
[0062]
t21.催化剂活化阶段:将原料混合液a升温至活化温度,加入催化剂,并充分混合,保温第一时长。
[0063]
一具体实施方式中,首先将原料混合物a升温值活化温度,例如所述活化温度为200℃-400℃,冷凝并收集气相,建立全回流。升温至目标温度后,加入催化剂mn
2+
盐,作为优选,催化剂的加入量为原料混合物a质量的1-1.5倍。充分搅拌,使得催化剂mn
2+
盐与原料混合物a充分混合,保温6h-24h。
[0064]
t22.缩合反应阶段:保温结束后,在第一反应温度下,滴加原料混合液a,进行缩合反应;冷凝收集气相馏分,得到粗品b。
[0065]
向催化剂mn
2+
盐保温活化结束后的反应体系中滴加原料混合液a,进行缩合反应。冷凝收集气相馏分,部分被作为回流液相返回反应体系,部分被采出,作为含2-甲基-1-[4-(叔丁基)苯基]-1-丙酮的粗品。作为优选,所述第一反应温度200℃-400℃。原料混合液a应被缓慢滴加进入反应体系内,作为优选,按每千克催化剂40g/h-75g/h的滴加速率,滴加原料混合液a,进行缩合反应。值得说明的是,所述“按每千克催化剂40g/h-75g/h的滴加速率,滴加原料混合液a”应该被理解为:如果体系内的催化剂含量为1kg,则原料混合液a的滴加速率为40g/h-75g/h;如果体系内的催化剂含量为2kg,则原料混合液a的滴加速率为80g/h-150g/h;如果体系内的催化剂含量为1000kg,则原料混合液a的滴加速率为40kg/h-75kg/h,以此类推。
[0066]
t23.分离阶段:从粗品b中分离未反应的原料异丁酸与叔丁基苯甲酸以及2-甲基-1-[4-(叔丁基)苯基]-1-丙酮。
[0067]
例如,采用蒸馏方法,从粗品b中分离原料异丁酸和中间产品2-甲基-1-[4-(叔丁基)苯基]-1-丙酮。
[0068]
t30.以2-甲基-1-[4-(叔丁基)苯基]-1-丙酮合成光引发剂185(2-羟基-2-甲基-1-[4-(叔丁基)苯基]-1-丙酮)。
[0069]
以下通过具体实验例,进一步说明本发明的技术方案以及技术效果。
[0070]
实验例一
[0071]
按照苯甲酸和异丁酸质量比为1.35:1、1.25:1、1.15:1,分别配置原料混合液a1、a2、a3。取2.5kg混酸及2kg催化剂mno加入到实验用釜式反应器中,进行搅拌加热活化。温度缓慢上升,待温度达到300℃时,开始按100g/h的滴加流量,向所述实验用釜式反应器中,滴加配置好的原料混合液a1、a2、a3,进行缩合反应。缩合反应产生的馏分,通过冷凝器进料冷凝收集,收集冷凝液进行精馏,多余的环己甲酸回收至混酸配置套用,得到中间体异丁酰苯,含量99%以上。得到的异丁酰苯通过氯化、碱解、精馏得到2-羟基-2-甲基-1-苯基-1-丙酮光引发剂。
[0072]
检测得到的异丁酰苯(未精馏前)的纯度,并计算异丁酰苯的收率,如表1。
[0073]
表1实验例一中得到的异丁酰苯的纯度和收率
[0074]
序号苯甲酸:异丁酸混酸投料量馏分重量纯度收率(按苯甲酸计算)a11.35:1200kg128.4kg98.2%90.37%a21.25:1200kg132.1kg99.5%97.41%a31.15:1200kg126.2kg99.2%96.36%
[0075]
由表1可以看出,mno催化作用下,得到的异丁酰苯的纯度能够达到99%以上,在异丁酸过量时,异丁酰苯的收率可以达到96%。同时,催化剂连续生产,至总投料量大于200kg,依然保持较高的催化活性。
[0076]
实验例二
[0077]
按照苯甲酸和异丁酸质量比为1.25:1配置原料混合液a4。取2.5kg混酸及2kg催化剂mnco3加入到实验用釜式反应器中,进行搅拌加热活化。温度缓慢上升,待温度达到320℃时,开始按80g/h的滴加流量,向所述实验用釜式反应器中,滴加配置好的原料混合液a4,进行缩合反应。缩合反应产生的馏分,通过冷凝器进料冷凝收集,收集冷凝液进行精馏,多余的环己甲酸回收至混酸配置套用,得到中间体异丁酰苯,含量99%以上。得到的异丁酰苯通过氯化、碱解、精馏得到2-羟基-2-甲基-1-苯基-1-丙酮光引发剂。
[0078]
检测得到的异丁酰苯(未精馏前)的纯度,并计算异丁酰苯的收率,如表2。
[0079]
实验例三
[0080]
按照苯甲酸和异丁酸质量比为1.25:1配置原料混合液a4。取2.5kg混酸及2kg催化剂苯甲酸锰(ⅱ)加入到实验用釜式反应器中,进行搅拌加热活化。温度缓慢上升,待温度达到280℃时,开始按80g/h的滴加流量,向所述实验用釜式反应器中,滴加配置好的原料混合液a4,进行缩合反应。缩合反应产生的馏分,通过冷凝器进料冷凝收集,收集冷凝液进行精馏,多余的环己甲酸回收至混酸配置套用,得到中间体异丁酰苯,含量99%以上。得到的异丁酰苯通过氯化、碱解、精馏得到2-羟基-2-甲基-1-苯基-1-丙酮光引发剂。
[0081]
检测得到的异丁酰苯(未精馏前)的纯度,并计算异丁酰苯的收率,如表2。
[0082]
表2实验例二、三中得到的异丁酰苯的纯度和收率
[0083]
序号苯甲酸:异丁酸混酸投料量馏分重量纯度收率(按苯甲酸计算)a41.25:1200kg133.7kg99.4%97.58%a41.25:1200kg134.3kg99.0%97.23%
[0084]
由表2可以看出,mnco3和苯甲酸锰(ⅱ)催化作用下,得到的异丁酰苯的纯度能够达到99%以上,在异丁酸过量时,异丁酰苯的收率可以达到97%以上,尤其地,苯甲酸锰(ⅱ)催化作用下,异丁酰苯的收率可以达到97%以上。同时,催化剂连续生产,至总投料量大于200kg,依然保持较高的催化活性。
[0085]
实验例四
[0086]
按照苯甲酸和异丁酸质量比为1.25:1配置原料混合液a4。取2500kg混酸及2000kg催化剂mno(g1)、mnco3(g2)、苯甲酸锰(ⅱ)(g3)、mno和mnco3的混合物(物质的量的比1:1)(g4)加入到容积为10m3的釜式反应器中,进行搅拌加热活化。温度缓慢上升,待温度达到300℃时,开始按100kg/h的滴加流量,向所述实验用釜式反应器中,滴加配置好的原料混合液a4,进行缩合反应。缩合反应产生的馏分,通过冷凝器进料冷凝收集,收集冷凝液进行精馏,多余的环己甲酸回收至混酸配置套用,得到中间体异丁酰苯,含量99%以上。
[0087]
统计当催化剂活性显著降低(采出的冷凝液中异丁酰苯的含量降低)时,装置连续
稳定运行的天数、产出的异丁酰苯的总量及反应釜釜底残液的量,计算生产单位质量异丁酰苯所产生的固体废弃物的量,如表3。
[0088]
表3实验例四的统计结果
[0089]
批号催化剂投料运行时间产出异丁酰苯釜残量吨产品产釜残量g12000kg404天632吨4200kg6.6kgg22000kg452天780吨4150kg5.3kgg32000kg420天720吨3820kg5.3kgg42000kg512天825吨4230kg5.1kg
[0090]
由表3可以看出,以mno(g1)、mnco3(g2)、苯甲酸锰(ⅱ)(g3)、mno和mnco3的混合物(物质的量的比1:1)(g4)作为催化剂,进行工业放大生产,催化剂的活性可维持釜式反应器连续稳定运行400天以上,生产每吨异丁酰苯所产生的固体废弃物的质量仅为5.1kg-6.6kg,是一种环境友好型生产工艺。
[0091]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1