一种三齿铕配合物、其制备方法及应用
1.本发明涉及稀土配合物、发光材料技术领域,具体涉及一种稀土铕配合物及其制备方法,以及在荧光免疫层析领域中的应用。
背景技术:
2.稀土配合物发光材料具有量子效率高、发光寿命长、stokes位移大、半峰宽窄,在显示、照明、免疫检测等领域中具有重要的应用前景。作为一种光致发光材料,由于有机配体具有很大的摩尔吸光系数,与纯无机稀土材料相比,配合物的吸光能力提高了上百倍。而且,稀土配合物相较于上转换稀土材料具有更高的量子效率、更低的毒性。更适合用于荧光免疫层析检测。
3.在稀土配合物中,有机配体对于配合物的发光性质起到了决定性的作用。有机配体与稀土离子螯合,不仅能够作为天线分子吸收和传递能量,而且能够保护稀土发光不被外界环境所淬灭。目前,大多数的稀土配合物以二齿配合物为主,但其光稳定性和热稳定性很差,在照明、显示以及免疫层析方面的应用存在缺陷。
4.为解决该问题,已有研究人员设计了三齿阴离子配体,通过增加配合物的螯合位点,当按3:1与稀土离子形成配合物后,就可以满足配位饱和的要求,避免了中性配体引入带来的热稳定性问题,从而得到热稳定性、光稳定性且高效的稀土配合物发光材料。但是由于阴离子配体的增加,稀土配合物的溶解性大幅度降低,影响了在免疫层析、生物成像等领域的应用。所以这类稀土配合物发光材料仍需做出必要的改进,以及使其具备更好的技术效果和更广泛的应用前景。
技术实现要素:
5.本发明是为了解决现有稀土配合物稳定性与溶解度无法同时满足的技术难题,获得一种稳定性好、同时溶解度高的稀土发光材料。从而更好的应用于荧光免疫层析等领域。
6.如前文所述,目前稀土配合物以二齿类为主,即使少量的三齿类也因其配体结构的刚性导致溶解度大幅度下降。为了在保证稀土配合物稳定性的前提下,提高其溶解度,本发明设计并使用新的合成方法合成了螯合基团,成功地改善了三齿稀土配合物的溶解度问题,并将其应用在了荧光免疫层析领域。
7.具体的,本发明的技术方案如下:
8.一种基于三齿4-羟基-1,5-萘啶类配体的稀土配合物,结构通式为eu(po)3,其中po代表磷氧基取代的4-羟基-1,5-萘啶类阴离子配体。配体po的结构如下所示:
[0009][0010]
其中,r1、r2、r3、r4为相同或不同的原子或基团,r1、r2、r3、r4各自独立地选自氢原子、卤素原子、烷基、烯基、炔基、芳香基、杂环基。
[0011]
优选地,上述卤素原子包括f、cl、br、i;烷基包括c1-c18的烷基,例如甲基、乙基、丙基、正丁基和异丁基;烯基包括c2-c18的烯基,例如乙烯、丙烯和丁烯;炔基包括c2-c18的炔基,例如乙炔、丙炔和丁炔;芳香基包括c6-c36的芳香基,例如苯基、苯甲酸和苯乙酮;杂环基包括式量在200以下的杂环基,例如噻吩、噻唑、呋喃和吡啶。
[0012]
其中r5为芳基。优选的,所述芳基为取代的苯基;进一步优选的,所述取代的苯基的取代基选自正丁基、正辛基、正十二烷基、正十六基、正十八烷基;优选正辛基、正十二烷基。
[0013]
上述配体po的合成路线如下:
[0014][0015]
优先地,配体po选自以下的任一种:
[0016][0017]
三齿铕配合物的结构如下所示:
[0018][0019]
优选地,三齿铕配合物的结构如下所示,这些结构具有较高的溶解度:
[0020][0021][0022]
本发明的三齿铕配合物的合成路线如下:
[0023][0024]
本发明的实施例还提供一种三齿铕配合物的合成方法,所述方法包括以下步骤:
[0025]
1)将原料2,2-二甲基-1,3-二恶烷-4,6-二酮,原甲酸三乙酯加入三口瓶中100 ℃反应1.5h,反应后再加3-溴-5-氨基吡啶,反应完毕。反应液趁热过滤,用乙醚洗涤得化合物a。
[0026]
2)将化合物a、二苯基醚加入到三口瓶中,回流反应1.5h,冷却至室温,过滤得固体产物,用二氯甲烷洗涤,得黑棕色产物b。
[0027]
3)将步骤2)得到的黑棕色产物b、碳酸钾加入到100ml的三口瓶中,加入 dmf,再加入苄氯85℃反应过夜。萃取,抽干dmf,柱层析得化合物c。
[0028]
4)将mg、i2加入到100ml的三口瓶中,再加入thf,将溶液加热。滴加4
‑ꢀ
丁基溴苯的thf溶液继续搅拌1h,然后冰浴至0℃,滴加二乙基氧磷的thf溶液 30min。室温搅拌2h。将反应用2m的hcl(20ml)淬灭,降温至0℃搅拌15min。用硅藻土过滤,滤液用二氯甲烷提取三次,柱层析得化合物d。
[0029]
5)将化合物c与化合物d加入到含k2co3的二氧六环溶液中,加入醋酸钯、二茂铁100℃回流过夜。萃取,柱层析得化合物e。
[0030]
6)将化合物e、二氧六环、浓盐酸加入到25ml的单口瓶中回流反应。用naoh溶液调成中性。用二氯甲烷萃取,柱层析得配体po。
[0031]
7)将po、naoh加入到单口瓶中,加入dmso室温搅拌直至澄清,回流10 min,滴加eucl3·
6h2o的乙醇溶液,70℃反应过夜。过滤得三齿铕配合物eu[po]3。
[0032]
在三齿铕配合物的制备中,主要难点与关键点在于三齿配体的合成。在发明专利申请公开cn105017329a中,北京大学团队公开了一种基于三齿阴离子配体的稀土铕配合物发光材料及其制备方法与应用,其中涉及三齿配体的合成并公开了三齿配体l1的合成路线及产率(三齿配体l1在cn105017329a专利申请公开中的编号为5f)。本发明在相关合成方法上进行了改进,获得了显著的有益效果:
[0033]
(1)发明专利cn105017329a的合成过程中需要用到活泼金属钠,该金属遇水易燃易爆,而本发明的合成步骤中不需要使用金属钠等危险化学药品,制备过程更加安全;
[0034]
(2)发明专利cn105017329a三齿配体l1的总产率为15%左右,而本发明采用新的合成方法使三齿配体的合成产率得到大幅改善,例如三齿配体l2的总产率在50%左右(三齿配体l2在本发明中的编号为po4)。
[0035]
本发明获得了一种稳定性好、溶解度高的稀土发光材料,适合应用于荧光免疫层析、生物成像、发光薄膜等领域。本发明示例性地将所合成的三齿铕配合物应用于免疫层析领域中。具体的,将铕配合物包覆在聚苯乙烯微球中,并对微球的表面进行修饰,最终将抗体偶联在微球表面,合成了荧光免疫微球。用于检测 saa项目,其检测灵敏度高,稳定性好,实现了高效快速的体外监测。
附图说明
[0036]
图1为配体po4的核磁氢谱图。
[0037]
图2为配体po4的核磁碳谱图。
[0038]
图3为配体po7的核磁氢谱图。
[0039]
图4为配体po7的核磁碳谱图。
[0040]
图5为三齿铕配合物eu[po]
3-4的激发和发射谱图。
[0041]
图6为三齿铕配合物eu[po]
3-4的热稳定性测试结果。
[0042]
图7为三齿铕配合物eu[po]
3-4的光稳定性测试结果。
[0043]
图8为结构修饰前后的三齿铕配合物的溶解度测试结果。
[0044]
图9为免疫层析微球的合成路线图。
[0045]
图10为荧光免疫层析检测示意图。
[0046]
图11为免疫层析检测标准曲线。
具体实施方式
[0047]
为了更加详细的说明本发明合成方案,下面将结合具体的合成方程式进行详细的介绍,显然所描述实施案例仅是本发明优先地一部分实施案例。在本研究领域内研究人员在没有作出任何的创新性贡献所提到的所有其他类似实施案例,都应当属于本发明保护的范围。
[0048]
本发明通过七步化学反应合成得到了稳定性好、溶解性好的三齿铕配合物。
[0049]
实施例1
[0050]
本实施的合成路线如下所示:
[0051][0052]
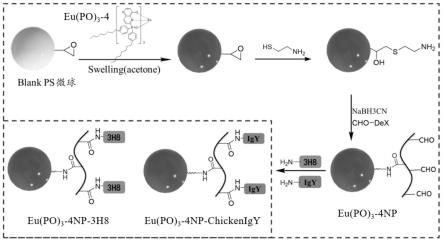
具体合成步骤如下:
[0053]
化合物a的合成:将2,2-二甲基-1,3-二恶烷-4,6-二酮(13.8g,96mmol,1.2eq),原甲酸三乙酯(55ml,320mmol,4eq)在100℃反应2h,反应后再加5-溴-3-氨基吡啶(13.76g,80mmol,1eq),继续反应。反应液趁热过滤,用石油醚洗涤得纯品化合物a。产率:95%。
[0054]
化合物b的合成:将化合物a(24g,72mmol)、二苯基醚(350ml)加入到 1000ml的三口瓶中,回流反应2h。冷却至室温,过滤得固体产物,用二氯甲烷洗涤,得黑棕色产物。然后用30eq的naoh水溶液搅拌一小时过滤得水溶液,用稀盐酸调至中性,过滤得固体产物化合物b。产率:50%。
[0055]
化合物c的合成:将化合物b(2g,9.5mmol)、碳酸钾(2g,15mmol)加入到100ml的三口瓶中,注射dmf(15ml),再注射苄氯(1.15ml,10mmol)85 ℃反应过夜。萃取,抽干dmf,柱层析得纯品化合物c。产率:70%。
[0056]
化合物d的合成:将mg(0.8g,33.3mmol,3.3eq),i2(5mg,0.02mmol)加入到100ml的三口瓶中,再加入thf(20ml),将溶液加热。滴加4-丁基溴苯(5.8 ml,30mmol,3eq)的thf溶液,继续搅拌1h,然后冰浴至0℃,滴加二乙基氧磷(1.3ml,10mmol,1.0eq)的thf溶液30min。室温搅拌2h。将反应用2m的hcl(20ml)淬灭,降温至0℃搅拌15min。用硅藻土过滤,滤液用二氯甲烷提取三次,柱层析得纯品化合物d。产率:70%。
[0057]
化合物e的合成:将化合物c(100mg,0.317mmol,1eq)与化合物d(150 mg,0.476mmol,1.5eq)加入到含k2co3(87mg,0.634mmol,2eq)的二氧六环溶液(5ml)中,与pd(oac)2(8.3mg,0.0317mmol 0.1eq),dppf(23mg 0.0138 mmol 0.1eq)100℃回流过夜。萃取,柱层析得化合物e。产率:80%。
[0058]
化合物po的合成,以三齿配体po4为例:将化合物e(50mg,0.09mmol)、 ch2cl2(2ml)、浓盐酸(1.13ml)加入到25ml的单口瓶中室温搅拌,回流反应,换二氧六环回流。用naoh溶液调成中性。用二氯甲烷萃取,柱层析得纯品化合物po4。产率:95%。
[0059]
化合物eu[po]
3-4的合成:将化合物po4(150mg,0.263mmol,3eq)加入到单口瓶中,
加入甲醇(3ml)室温搅拌,滴加eucl3·
6h2o(32mg,0.0876mmol,1 eq)的甲醇溶液(1ml),70℃反应过夜。过滤得纯品化合物eu[po]
3-4。产率: 90%。
[0060]
实施例2
[0061]
本实施例中的合成路线如下所示:
[0062][0063]
合成步骤同实施例1,只是r5不同。
[0064]
实施例3
[0065]
本实施例中的合成路线如下所示:
[0066][0067]
合成步骤同实施例1,只是r1、r2不同。
[0068]
实施例4
[0069]
本实施例中的合成路线如下所示:
[0070][0071]
合成步骤同实施例1,只是r2、r4、r5不同。
[0072]
实施例5
[0073]
本实施例中的合成路线如下所示:
[0074][0075]
合成步骤同实施例1,只是r1、r2、r4、r5不同。
[0076]
实施例6三齿配体po4的核磁谱图表征
[0077]1h nmr(400mhz,cdcl3)δ8.64(s,1h),8.35(s,1h),7.95(s,1h),7.65(s, 4h),7.20(s,4h),6.83(s,1h),2.57(t,j=7.3hz,4h),1.55(s,4h),1.26(m,j=8.2 hz,20h),0.87
(t,j=6.7hz,6h).
13
c nmr(101mhz,cdcl3)δ155.28,153.97, 148.15,142.97,139.28,137.45,132.10,129.12,128.85,128.12,127.06,111.72,35.93, 31.79,30.97,29.33,29.26,29.14,22.57,13.99.hrms for[m-pf6]
+
:calcd 1855.2734, found 1855.2965.
[0078]
三齿配体po4的核磁谱图表征如图1和图2所示。三齿配体po7的核磁谱图表征如图3和图4所示。由附图1和附图2的结果确认了三齿配体po4的结构;由附图 3和附图4的结果确认了三齿配体po7的结构。
[0079]
实施例7三齿铕配合物的热稳定性、光稳定性的表征
[0080]
本发明的技术效果主要通过实施例中的稀土配合物的发射光谱、热稳定性和紫外耐受性来体现。其中,紫外耐受性是通过测试稀土配合物掺杂的高分子薄膜(pmma)在紫外光照射下的发光强度衰减情况来衡量的。
[0081]
三齿铕配合物eu[po]
3-4的激发与发射光谱如图5所示。
[0082]
三齿铕配合物eu[po]
3-4的热稳定性测试如图6所示。
[0083]
三齿铕配合物eu[po]
3-4的光稳定性测试如图7所示。
[0084]
在热稳定性和光稳定性测试中,均采用两种常见的铕配合物作为对照。两种对照物分别为:eu(tta)3phen;eu(dbm)3phen。
[0085]
如图6所示,与两种常见的铕配合物对比,三齿铕配合物eu[po]
3-4的热稳定性明显增强。如图7所示,与两种常见的铕配合物对比,三齿铕配合物eu[po]
3-4 的光稳定性明显增强。
[0086][0087]
实施例8三齿铕配合物的溶解度
[0088]
使用丙酮、甲苯等试剂对eu[po]
3-4、eu[po]
3-7配合物进行了溶解度测试。本发明提出的三齿铕配合物经过结构修饰,其溶解度得到大大提高。如图8所示,三齿配体r5苯环上碳链的c原子个数n=8时为eu[po]
3-4;三齿配体r5苯环上碳链的c原子个数n=4时为eu[po]
3-7;三齿配体r5苯环上碳链的c原子个数n=0时为发明专利cn105017329a中公开的铕配合物eu5,不修饰长碳链,作为对比其溶解度较小。
[0089]
实施例9三齿铕配合物包覆在微球中制备荧光微球
[0090]
取聚苯乙烯微球(1.185ml)、去离子水(2.82ml)、丙酮(5ml)加入反应瓶中,室温搅拌20min。加入eu[po]
3-4三齿铕配合物的丙酮溶液(1ml,20 mg/ml)继续室温搅拌5h。反应后离心(12000r/min,10min),加入去离子水 /乙醇(v:v=9:1)反复洗3次。加入10ml去离子水储存。固含量1%。
[0091]
实施例10氨基荧光微球的制备
[0092]
取实施例9所得到的荧光微球(20ml,10mg/ml)离心(12000r/min,10min) 去除上清液,加入naoh水溶液(20ml,ph=11),超声(10min)。称取巯基乙胺(30mg)一次性加入上述微球溶液中,震荡后室温反应24h。反应后离心 (8000r/min,10min),加入水/乙醇(9:1)反
复离心、洗3次。加入20ml bbs (ph=7.5)储存,固含量1%。
[0093]
实施例11醛基荧光微球的制备
[0094]
取实施例10所得到的含氨基荧光微球的mes溶液(ph=6.0)(15ml,10 mg/ml)加入醛基葡聚糖(50万,1.25mmol/g,100mg)反应20min后,加入氰基硼氢化钠(100μl,50mg/ml),继续反应过夜。离心(7000r/min,5min),加入去离子水反复洗三次。加入15ml bbs(ph=7.5)储存。固含量1%。所得的微球命名为eu[po]
3-4np,为表面含有醛基的荧光微球。
[0095]
实施例12免疫微球的制备
[0096]
取实施例11所得到的eu[po]
3-4np醛基荧光微球,将醛基荧光微球(625μl, 10mg/ml)加入bbs溶液(3ml,ph=7.5)。将鸡igy(5mg/ml,200μl)加入上述微球中反应20min后加入氰基硼氢化钠(20μl,50mg/ml),室温反应6h。然后离心(7000r/min,5min)移除上清液,加入4ml bbs缓冲溶液(ph=7.5),超声5min,bsa(300μl,10%)封闭1h,离心、加5ml bbs,固含量0.25%。
[0097]
实施例13免疫层析试纸条的制备
[0098]
免疫层析试纸条由pvc塑料底板、nc膜、样品垫和吸水纸四部分组成。制备方法如下:
①
将nc膜粘贴在pvc塑料底板上。
②
配制saa抗体5f2蛋白浓度为1.5mg/ml以1μl/cm的速度划在nc膜上作为t线;配制羊抗鸡igy蛋白浓度为1 mg/ml以1μl/cm的速度划在nc膜上作为c线。
③
使用喷金稀释液配制eu[po]3‑ꢀ
4np-3h8免疫微球和eu[po]
3-4np-chicken igy浓度为25μg/ml和5μg/ml,使用 xyz三维划膜喷金仪将上述稀释液喷在样品板上,其中5f2和3h8免疫配对。
④
将划有t/c线的nc膜和喷有免疫微球的样品板放在37℃的恒温箱中过夜。
⑤
将样品垫和吸水纸分别粘贴在nc膜的固定位置。
⑥
使用微电脑自动斩切仪将试纸条剪切为宽度为0.38cm,并将其安装在塑料卡槽中,干燥常温环境下储存备用。
[0099]
实施例14saa项目检测
[0100]
以正常人的血清为样品稀释液,将saa标准品稀释为0μg/ml、0.5μg/ml、1μg/ml、2μg/ml、5μg/ml、10μg/ml、20μg/ml、55μg/ml、110μg/ml、220 μg/ml、440μg/ml,各取不同浓度的标准品1.25μl和98.75μl saa样品稀释液混匀并加入到加样口中,反应11min后检测、记录t线和c线的荧光强度并计算 i
t
/ic值,检测重复三次,绘制saa标准品浓度与对应i
t
/ic值的曲线(结果如图11 所示)。
[0101]
本发明制备的三齿铕配合物具有较好的稳定性、较好的溶解度,并将其应用于荧光免疫层析中,获得了一个高灵敏度的荧光免疫试纸检测卡,用于检测saa 项目,由附图11可知,其检测限低至2μg/ml,低于临床值10μg/ml,检测浓度高达220μg/ml。
[0102]
表1免疫层析平台用于检测saa的分析评价结果
[0103][0104]
实施例14三齿配体合成方法
[0105]
在三齿铕配合物的制备中,主要难点与关键点在于三齿配体的合成。
[0106]
在发明专利申请公开cn105017329a中,北京大学团队公开了一种基于三齿阴离子配体的稀土铕配合物发光材料及其制备方法与应用,其中涉及三齿配体的合成并公开了三齿配体l1的合成路线及产率(三齿配体l1在cn105017329a专利公开中的编号为5f)。
[0107][0108]
本发明在相关合成方法上进行了改进,获得了显著的有益效果:
[0109]
(1)发明专利cn105017329a的合成过程中需要用到活泼金属钠,该金属遇水易燃易爆,而本发明的合成步骤中不需要使用金属钠等危险化学药品,制备过程更加安全;
[0110]
(2)发明专利cn105017329a三齿配体l1的总产率为15%左右,而本发明采用新的合成方法使三齿配体的合成产率得到大幅改善,例如三齿配体l2的总产率在50%左右(三齿配体l2在本发明中的编号为po4)。
[0111]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1