siRNA在制备由G6PD高表达导致细胞恶性转化的疾病的药物中的应用
sirna在制备由g6pd高表达导致细胞恶性转化的疾病的药物中的应用
技术领域
1.本发明属于生物医药技术领域,尤其是指一种sirna在制备由g6pd 高表达导致细胞恶性转化的疾病药物中的应用。
背景技术:
2.无机砷化合物是iarc确认的人类致癌物,但其致癌的分子机制研究长期滞后。目前,对砷致癌机制的研究主要集中在氧化应激,已被广泛关注与接受,而对还原应激重视不够。然而,氧化和还原反应伴随大多数的细胞生物学过程,在砷致细胞恶性转化过程中除了关注过度氧化反应导致的氧化应激,还应关注过度还原反应导致的还原应激,精准观察细胞氧化还原状态。研究发现,不同细胞器中的氧化还原状态是不同的。因此,除了关注细胞的氧化还原水平外,还应该关注不同细胞器的氧化还原状态。越来越多的研究发现,砷对线粒体具有明显的毒性作用。本课题组也发现naaso2在诱导细胞恶性转化的过程中,细胞线粒体dna拷贝数明显增加,电子显微镜观察到细胞内线粒体肿胀。线粒体是ros产生的主要来源,砷及其化合物能够通过线粒体产生ros,导致氧化应激和线粒体的功能异常。线粒体电子传输链也是atp合成的主要来源,线粒体基质中较高水平的nadh/nad
+
对于线粒体atp产生的还原力是必需的。通过调节三羧酸循环相关的酶来调节新陈代谢,因此线粒体的氧化还原状态参与细胞代谢的调节。氧化还原调节与细胞稳态密切相关,正常生理情况时,人体内氧化还原处于稳态平衡,为生物大分子发挥其生理功能提供了一个稳定的微环境。但当机体受到刺激时,体内氧化与还原系统间的平衡被打破。为此急需一种能实现体内动态平衡的药物。
3.那么,是什么原因引起的氧化还原稳态失衡?细胞氧化还原状态是细胞内偶联酶系统和小分子抗氧化剂(还原剂)组成的抗氧化/氧化还原网络,以防范氧化剂造成的潜在损害,保持细胞功能和健康。还原应激是指机体的电子压力或还原当量的异常升高,如公认的细胞内主要的氧化还原对还原型谷胱甘肽/氧化型谷胱甘肽(gsh/gssg)和还原型辅酶ii/氧化型辅酶ii (nadph/nadp
+
)等的升高,而机体不能有效缓解这种电子压力而引起细胞内氧化还原状态失衡的一种状态。nadph/nadp
+
和gsh/gssg产生的主要来源是葡萄糖代谢过程中的磷酸戊糖途径(ppp),nadp
+
在葡萄糖-6
‑ꢀ
磷酸脱氢酶(g6pd)和6-磷酸葡萄糖脱氢酶(pgd)作用下生成nadph,而nadph使gssg在氧化还原循环酶、谷胱甘肽还原酶作用下生成gsh。研究发现,g6pd介导的磷酸戊糖途径增强能够增加细胞内核苷酸的合成,增强细胞抗氧化防御能力,从而促进细胞增殖。而磷酸戊糖途径增强是癌细胞发生代谢重组的特点之一,为了满足细胞不断增殖的需求,癌细胞通过增强磷酸戊糖途径等关键代谢途径重组代谢,从而增强癌细胞的侵袭能力和转移特性。
技术实现要素:
4.为解决以上技术问题,本发明提供了一种g6pd sirna在制备由g6pd 高表达导致
细胞恶性转化的疾病的药物中的应用。
5.本发明的第一个目的在于提供一种g6pd sirna在制备由g6pd高表达导致细胞恶性转化的疾病药物中的应用。其中g6pd sirna选自sc-60667a、 sc-60667b或sc-60667c中的一种或多种。
6.sc-60667a:
7.正义链:5
′‑
gaaggucaagguguugaaatt-3
′
8.反义链:5
′‑
uuucaacaccuugaccuuctt-3
′
9.sc-60667b:
10.正义链:5
′‑
gcaacagauacaagaacgutt-3
′
11.反义链:5
′‑
acguucuuguaucuguugctt-3
′
12.sc-60667c:
13.正义链:5
′‑
gaguggguuuccaguaugatt-3
′
14.反义链:5
′‑
ucauacuggaaacccacuctt-3
′
。
15.在本发明的一个实施例中,所述细胞选自hacat。
16.在本发明的一个实施例中,所述疾病为皮肤基底细胞癌或皮肤鳞状细胞癌。
17.本发明的第二个目的在于提供一种重组载体,包括能够转录所述g6pdsirna的序列,所述序列嵌入载体。
18.本发明的第三个目的在于提供一种药物组合物,包括任一项所述的药物或所述的重组载体。
19.在本发明的一个实施例中,还包括药学上或药理上可接受的载体。
20.在本发明的一个实施例中,所述载体选自崩解剂、稀释剂、润滑剂、粘合剂、湿润剂、矫味剂、助悬剂、表面活性剂和防腐剂中的一种或多种。
21.在本发明的一个实施例中,药物组合物的剂型为片剂、胶囊剂、软胶囊剂、颗粒剂、丸剂、口服液、乳剂、干混悬剂、干浸膏剂或注射剂。
22.本发明的第四个目的在于提供一种试剂盒,包括所述重组载体、所述的药物组合物。
23.本发明的第五个目的在于提供所述的试剂盒在制备治疗由g6pd高表达导致细胞恶性转化的疾病的药物中的应用。
24.在本发明的一个实施例中,所述细胞选自hacat。
25.在本发明的一个实施例中,所述疾病为皮肤基底细胞癌或皮肤鳞状细胞癌。
26.本发明的上述技术方案相比现有技术具有以下优点:
27.本发明对线粒体的氧化还原状态检测发现线粒体中nadph/nadp
+
和 gsh/gssg水平显著升高。进一步构建荧光标记线粒体谷胱甘肽的mito-grx1-rogfp2 hacat细胞株,1.0μm naaso2连续处理 mito-grx1-rogfp2 hacat细胞中线粒体gsh/gssg水平与细胞中水平一致,也显著升高,表明细胞线粒体还原水平增加。
28.本发明发现naaso2致hacat细胞恶性转化前期,细胞呈现正常的代谢方式,即以三羧酸循环为主;而恶性转化后期,葡萄糖6磷酸水平,乳酸分泌水平,g6pd水平均显著升高;乙酰辅酶a水平显著降低。检测代谢重组相关关键酶的表达水平发现,naaso2处理组细胞的hk-2、pfkfb3、pdk1 以及pgd和g6pd表达水平均明显增高,表明naaso2致hacat细胞恶性转
60667:g6pd sirna包括三种不同的sirna,其中三种sirna的序列:
45.sc-60667a:
46.正义链:5
′‑
gaaggucaagguguugaaatt-3
′
47.反义链:5
′‑
uuucaacaccuugaccuuctt-3
′
48.sc-60667b:
49.正义链:5
′‑
gcaacagauacaagaacgutt-3
′
50.反义链:5
′‑
acguucuuguaucuguugctt-3
′
51.sc-60667c:
52.正义链:5
′‑
gaguggguuuccaguaugatt-3
′
53.反义链:5
′‑
ucauacuggaaacccacuctt-3
′
。
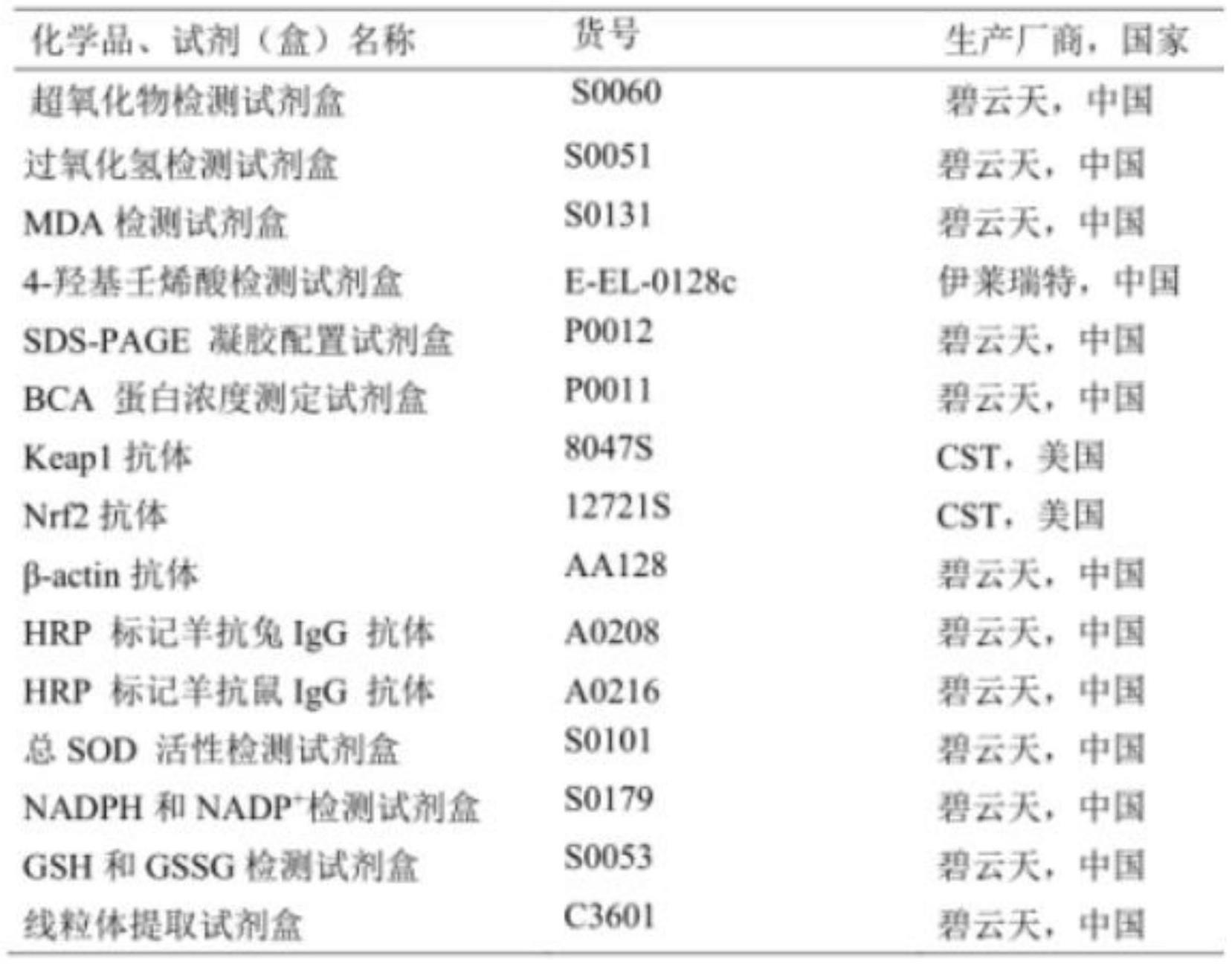
54.4,所用试剂盒:
[0055][0056]
实施例1细胞培养及染砷
[0057]
用dmem完全培养基培养细胞,放置于含5%co2、37℃恒温培养箱中。细胞传代时,将细胞培养皿中的培养基倒掉,加入pbs清洗细胞2次,向培养皿中添加1.5ml含edta的胰酶消化液,并置于37℃培养箱中消化6 min,镜下观察当细胞形态发生变化并脱离细胞培养皿表面呈漂浮状态时,再添加2ml培养基以终止消化,将细胞悬液转移到到无菌离心管中离心 (1000g室温离心3min),尽量吸除上清,保留细胞沉淀,添加2ml培养基并使用移液器轻柔吹散细胞,将重悬细胞按照1:2传代培养。用含1.0μm naaso2的dmem培养基培养到35代(约18周),同时设立未染砷的传代对照组细胞。
[0058]
实施例2 g6pd sirna处理恶性转化的hacat细胞
[0059]
培养恶性转化的hacat细胞和正常传代对照组细胞于6孔板中,细胞贴壁后处理。
配制sirna转染试剂,每培养皿配置溶液a:12μl g6pd sirna 或con sirna,加入988μl sirna转染介质。每培养皿配制溶液b:12μl sirna转染试剂,加入988μl sirna转染介质。a、b溶液充分混合后得到含有转染试剂和g6pd sirna或con sirna的混合物,避光孵育20min。细胞先用pbs清洗一次,然后用2ml sirna转染介质清洗一次,吸出液体,弃掉液体。每培养皿加入2ml sirna转染试剂混合物,置于含5%co2、37 ℃恒温培养箱中孵育细胞6h。吸出培养皿内的液体,加入适量正常细胞培养基,于24-72h后检测相关指标。
[0060]
如图1所示,与传代对照细胞(未经naaso2处理的细胞)相比,1.0μm naaso2所致恶性转化的hacat(t-hacat)细胞g6pd表达水平显著增高 (p《0.05)。t-hacat细胞转染g6pd sirna后:与转染对照组(sirna con) 细胞相比,g6pd表达水平明显降低(p《0.05),表明转染成功。
[0061]
实施例3 t-hacat细胞中nadp
+
/nadph和gsh/gssg的检测
[0062]
具体实验操作:
[0063]
(一),t-hacat细胞中nadp
+
/nadph:
[0064]
a.细胞样品的准备:1
×
106个细胞中加入200μl的nadp
+
/nadph提取液,用移液枪充分吹打后置于冰上。12,000g,4℃离心10分钟,取上清待测。
[0065]
2)试剂盒的准备工作:
[0066]
a.按照说明书要求用双蒸水配制1mm nadph标准品。分装后-80℃避光保存。
[0067]
b.nadph标准曲线的设置:把1mm的nadph标准品用提取液稀释成相应浓度。
[0068]
c.g6pdh工作液的配制:根据样品的数量,取适量的g6pdh用反应缓冲液稀释50倍。每个样品需使用100μl的g6pdh工作液。
[0069]
3)样品测定:
[0070]
a.样品中nadp
+
/nadph总量的测定:在96孔板中加入50μl待测样品。
[0071]
b.样品中nadph的含量测定:吸取200μl待测样品于离心管中,pcr 仪上60℃加热30min以分解nadp
+
,检测得到的是样品中nadph的浓度。
[0072]
c.请参考下表进行加样。轻柔加入g6pdh工作液,以免产生气泡,充分混匀。若不慎出现气泡,使用细小的针头戳破。
[0073][0074]
d.在450nm处测量吸光度值。
[0075]
4)样品中nadp
+
/nadph量的计算:
[0076]
a.标准品的吸光度=标准品组中每个点的平均吸光度-空白对照组的吸光度。
[0077]
b.根据标曲计算nadp
+
和nadph总浓度或者nadph的浓度。
[0078]
c.根据如下计算公式,计算nadp
+
的量以及nadp
+
/nadph的比值。
[0079]
[nadp
+
]=[nadp total]-[nadph]
[0080]
[nadp
+
]/[nadph]=([nadp total]-[nadph])/[nadph],实验结果见图4。
[0081]
(二),细胞中gsh的检测:
[0082]
1)按照试剂盒的说明配制好以下工作液:gssg储备液(5mg gssg加入816μl水)、dtnb储备液(4.5mg dtnb加入1.5ml dmso)、蛋白去除试剂m溶液(0.05g蛋白去除试剂m加入1ml检测缓冲液)、40mg/mlnadph储备液(4mg nadph加入100μl水)、总谷胱甘肽检测工作液、5 倍稀释谷胱甘肽还原酶(5μl谷胱甘肽还原酶加入200μl缓冲液)、0.5mg/mlnadph(10μlnadph加入790μl缓冲液)、gsh清除试剂工作液(取10.8 μl加入89.2μl无水乙醇)和稀释gsh清除辅助液(取53ul清除辅助液加入47μl水)。
[0083]
2)培养细胞,用pbs洗一次,1,000g离心收集细胞。每10μl细胞沉淀,加入30μl蛋白去除试剂m溶液,涡旋混匀。用低温液氮和37℃水浴对样品进行快速冻融两次。冰浴5min后4℃、10,000g离心10min。收集上清,测定总谷胱甘肽的含量。
[0084]
3)测定样本中gssg含量:取部分上述样品,加入稀释的gsh清除辅助液和清除试剂工作液,参照下表。检测结果见图2和图3。
[0085][0086]
如图2所示,检测nadph/nadp
+
比值发现,与传代对照细胞相比, t-hacat细胞的nadph/nadp
+
水平增高(p《0.05)。对t-hacat细胞转染g6pd sirna之后发现,与转染对照组相比,nadph/nadp
+
比值明显降低(p《0.05)。如图3所示,检测gsh/gssg比值发现,与传代对照细胞相比,t-hacat细胞的gsh/gssg水平增高(p《0.05)。对t-hacat细胞转染g6pd sirna之后发现,与转染对照组细胞相比,gsh/gssg比值显著降低(p《0.05)。表明g6pd sirna能够逆转1.0μmnaaso2所致t-hacat 细胞氧化还原水平失衡。
[0087]
实施例4 t-hacat细胞中线粒体nadph/nadp
+
和gsh/gssg水平检测
[0088]
具体实验操作:
[0089]
t-hacat线粒体提取:1)常温融化试剂盒中的溶液后置于冰上(不需要加入pmsf)。
[0090]
2)培养细胞,用pbs洗一次,胰酶消化后1,000g离心收集细胞。
[0091]
3)用冷的pbs轻轻重悬,取少量进行细胞计数,1,000g离心收集剩余细胞。
[0092]
4)加入2ml分离试剂,重悬后,冰浴15min。
[0093]
5)用玻璃匀浆器匀浆细胞悬液15下。
[0094]
6)600g,4℃离心10min后留取上清。
[0095]
7)11,000g,4℃离心10min后弃上清,沉淀即为分离的线粒体。线粒体中nadph与gsh检测方法同实例3中的检测方法。实验结果见图4和图5。
[0096]
如图4所示,检测线粒体nadph/nadp
+
比值发现,与传代对照细胞相比,t-hacat的
线粒体nadph/nadp
+
水平显著增高(p《0.05)。对t-hacat 细胞转染g6pd sirna之后发现,与转染对照组细胞相比,线粒体 nadph/nadp
+
比值降低(p《0.05)。如图5所示,检测线粒体gsh/gssg 比值发现,与传代对照细胞相比,t-hacat细胞的线粒体gsh/gssg水平明显增高(p《0.05)。对t-hacat细胞转染g6pd sirna之后发现,与转染对照组细胞相比,线粒体gsh/gssg比值降低(p《0.05)。上述实验结果表明,g6pd sirna能够逆转1.0μm naaso2所致t-hacat细胞线粒体氧化还原水平失衡。
[0097]
实施例5 mito-grx1-rogfp2 t-hacat中线粒体gsh/gssg水平检测
[0098]
培养细胞,用酶标仪在发射波长为525nm,激发波长分别为405nm和 488nm时,检测488/405nm的比值即表示:gsh(reduced glutathione)/gssg (oxidized glutathione)的比值。实验结果见图6。
[0099]
如图6所示,检测线粒体gsh/gssg比值发现,与传代对照组细胞相比,1.0μm naaso2恶性转化mito-grx1-rogfp2 hacat(mito-grx1-rogfp2t-hacat)细胞的线粒体gsh/gssg水平明显增高(p《0.05)。对恶性转化细胞转染g6pd sirna之后,与转染对照组细胞相比,线粒体gsh/gssg 比值明显降低(p《0.05)。上述实验结果表明,g6pd sirna能够逆转1.0μmnaaso2所致mito-grx1-rogfp2 t-hacat细胞线粒体gsh/gssg比值的升高,导致线粒体还原水平降低。
[0100]
实施例6细胞倍增时间检测
[0101]
具体实验操作:用胰酶消化收集细胞,按照每孔10,000个接种于24孔板中。然后每隔24h,每组收取3孔细胞分别计数,根据公式计算细胞数量增加一倍所需要的时间:(t
×
lg2)/(lg nh-lg ni)。t为培养时间,nh为培养t时间(h)后的细胞数,ni为初始接种细胞数。实验结果见图7。
[0102]
如图7所示,检测细胞倍增时间发现,与传代对照组细胞相比,t-hacat 细胞倍增时间明显降低(p《0.05)。对t-hacat细胞转染g6pd sirna之后,与转染对照组细胞相比,细胞倍增时间明显升高。
[0103]
实施例7细胞划痕实验
[0104]
具体实验操作:1)消化收集细胞接种于六孔板中,置于细胞培养箱中培养。
[0105]
2)培养约24h待细胞长满,用200μl枪头垂直于板孔划痕,尽可能使划痕宽度一致。
[0106]
3)用移液枪将旧培养基吸去,pbs冲洗细胞3次后,加入无血清培养基,拍照后继续培养。
[0107]
4)48h后,再次拍照,每孔至少选取3个划痕视野进行拍照,用imagej软件进行定量分析,实验结果见图8。
[0108]
如图8所示,划痕实验发现,与传代对照细胞组相比,t-hacat细胞的细胞迁移能力明显增强(p《0.05)。对t-hacat细胞转染g6pd sirna之后,与转染对照组细胞相比,细胞迁移能力明显降低。
[0109]
实施例8软琼脂克隆形成实验
[0110]
具体实验操作:1)在双蒸水中加入0.504g低熔点琼脂糖,得到浓度为 1.4%的琼脂糖溶液,经高压灭菌后42℃水浴,与同体积的2
×
dmem完全培养基混合,得到浓度为0.7%的琼脂糖溶液。用移液枪吸1.5ml 0.7%的琼脂糖溶液缓缓加入到35mm培养皿皿底,室温凝固后即为底层胶。
[0111]
2)细胞进行计数后,调整细胞浓度,将5,000个细胞置于dmem完全培养基中加入等体积的浓度为0.7%的琼脂糖溶液,得到琼脂糖浓度为0.35%的细胞悬液,用移液枪吸1.5ml加到底层胶上面得到顶层胶,待其凝固后加入1.0ml dmem完全培养基,于5%co2、37℃连续培养4周,每2到 3天换液一次。在显微镜下,选取细胞直径大于50μm的克隆集落进行计数。实验结果见图9。
[0112]
如图9所示,与传代对照组细胞相比,t-hacat细胞的锚着独立生长能力显著增强(p《0.05)。对t-hacat细胞转染g6pd sirna之后,与转染对照组细胞相比,细胞锚着独立生长能力显著降低(p《0.05)。上述实验结果表明,g6pd sirna能够逆转1.0μm naaso2所致t-hacat细胞恶性转化表型。
[0113]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1