一种在Raw264.7细胞中快速构建稳定高表达细胞株的方法与流程
一种在raw264.7细胞中快速构建稳定高表达细胞株的方法
技术领域
1.本发明涉及分子生物技术及基因工程领域,尤其是一种在raw264.7细胞中快速构建稳定高表达细胞株的方法。
背景技术:
2.腹膜透析对于终末期肾病患者来说是一种成功的肾脏替代疗法,而腹膜炎是导致腹膜透析失败甚至患者死亡的主要原因,外界病原体的侵入,诱导巨噬细胞及白细胞等侵入腹膜。
3.巨噬细胞是属于吞噬细胞的一种细胞,是人体免疫系统的主要成员之一,能识别不同的病原微生物,并且能够快速做出有效反应,起到清除病原体的作用,是机体抵御外来感染的第一道防线。
4.pparγ即过氧化物酶体增殖物活化受体γ,是一种核激素受体超家族中的脂肪酸激活转录因子,经研究,pparγ在许多炎症疾病包括腹膜炎,脑损伤中的炎症,肺炎,肠炎,乳腺炎等许多不同部位的炎症中,pparγ都起着抗炎的作用。因此,通过研究pparγ是否以外泌体的形式从巨噬细胞中分泌出来,以及进一步研究分泌的pparγ调控腹膜炎性损伤的机制研究能够从机体防御的角度,为腹膜炎的预防及治疗提供新的思路。
5.在该研究过程中,在巨噬细胞raw264.7细胞中构建pparγ稳定高表达的细胞株是实验开展的基础,然而,慢病毒感染raw264.7十分困难,因此,提供一种在raw264.7细胞中快速构建稳定高表达pparγ的细胞株的方法并进行应用是需要解决的一个技术问题。
技术实现要素:
6.本发明所要解决的技术问题是提供一种在raw264.7细胞中快速构建稳定高表达细胞株的方法,解决了raw264.7难感染的问题。
7.为解决上述技术问题,本发明的技术方案是:一种在raw264.7细胞中快速构建稳定高表达细胞株的方法,包括以下步骤:(1)将目的基因与载体连接,转化到感受态dh5α;(2)对转化后的dh5α扩大培养,提取质粒;(3)将获得的质粒转染293t细胞,获得慢病毒液;(4)利用ptg8000浓缩病毒液的方法浓缩病毒液;(5)利用电转仪通过电转的方式将浓缩病毒转入raw264.7细胞;(6)稳定表达细胞的筛选;(7)细胞扩增;(8)获得稳定高表达目的基因的raw264.7细胞株。
8.慢病毒感染并非平常的感染,而是将包装好的慢病毒颗粒在借助外力的作用下(电转仪),通过高强度的电场作用,瞬时提高细胞膜的通透性,从而吸收周围介质中的病毒颗粒,实现构建稳定高表达目的基因的细胞株。本发明其能够实现目标基因pparγ的稳定
过表达,进而为在raw264.7细胞中研究pparγ的分泌,以及分泌的pparγ调控腹膜炎性损伤的机制研究提供了实验材料,且该细胞株过表达pparγ稳定,可以用于定量和定性检测pparγ过表达状态下相关蛋白质的表达情况。
9.作为改进,所述raw264.7细胞为宿主细胞。
10.作为改进,述步骤(3)的具体步骤包括:a)转染前24h用胰蛋白酶消化对数生长期的293t细胞,接种于10cm大皿中,37℃、5%co2培养箱内用10%血清的dmem培养基培养;b)取2个无菌1.5ml离心管置于离心管架分别a管和b管,取650ulopti-mem 加入到两个离心管,取36μl lipofectamine 2000于a管中,分别取8ug质粒 3*flag-pparγ-egfp、6μgplp1、6μg plp2、6μg vsvg 加入b管中,室温静置5min;c)将长至80%的293t培养基弃掉,pbs清洗后换成opti-mem培养基培养;d)5min后,将a、b两管混合,室温静置20min;e)将a、b混合液转移至293t细胞的培养液中,混匀,于37℃、5% co2细胞培养箱中培养;f)培养6 h后弃去含有a、b混和物的培养基, pbs清洗,加入含10%灭活血清的dmem细胞培养基6 ml,于37℃、5% co2培养箱内继续培养48 h;g)收集上清,1000 rpm离心5min,弃去细胞碎片,上清液用0.45 μm pvdf滤器过滤至5 ml圆底离心管中。
11.作为改进,所述步骤(5)的具体步骤包括:a)将长至70%-80%的两皿raw264.7细胞拿到安全柜中,吹打下细胞,收集细胞沉淀,230g,3min离心;b)弃上清,pbs清洗一遍,分装为两管,230g,3min离心,弃上清;c)电转缓冲液为pbs和灭活的胎牛血清,用1ml血清/pbs重悬,计数,取5
×
106 个细胞于10 μl病毒浓缩液中吹打均匀,使用电极杯配套吸管加入到2mmbtx电极杯中,设置电压为200v,脉冲长度为8毫秒,脉冲数量为1,脉冲场强为1000v/cm,总样本体积为200
µ
l,温度为室温;d)将电转后的细胞悬液吸出加入含有1ml灭活的胎牛血清的六孔板中,37℃、5% co2培养箱内中培养。
12.本发明与现有技术相比所带来的有益效果是:本发明提供的方法通过利用电转仪,通过电转的方式将浓缩病毒转入raw264.7细胞,从而攻克了raw264.7难感染的难题,从而能够快速构建稳定高表达目标基因的raw264.7细胞株,能够通过western blot、免疫荧光等操作进行相关蛋白的定量和定性。
附图说明
13.图1为western blot实验结果图i。
14.图2为western blot实验结果图ii。
15.图3为过表达pparγ后炎症因子下调的实时荧光定量pcr结果图。
具体实施方式
16.下面结合说明书附图对本发明作进一步说明。
17.一种在raw264.7细胞中快速构建稳定高表达细胞株的方法,包括以下步骤:(1)将目的基因与载体连接,转化到感受态dh5α;(2)对转化后的dh5α扩大培养,提取质粒;对转化后dh5α进行质粒提取,获得3*flag-pparγ-egfp质粒;(3)将获得的质粒转染293t细胞,获得慢病毒液;以所述3*flag-pparγ-egfp质粒转染293t细胞,收获病毒液,并以所述病毒液使用peg8000浓缩方法浓缩,利用电转仪感染raw264.7细胞后筛选出阳性克隆,培养10天左右,获得稳定高表达pparγ的细胞株,具体步骤如下:a)转染前24h,观察293t细胞状态,长至90%左右,状态良好;弃旧培养液,加入1ml pbs溶液,缓慢晃动,洗涤细胞,弃去pbs溶液,每皿加入1ml胰酶,消化约30s直到细胞变圆,变亮,弃胰酶,加入2ml ldmem+10%fbs培养基,用枪吹打数次,将皿底的细胞冲洗下来,收集到5ml离心管中, 230g,3min离心;加入1ml dmem+10%fbs培养基重选均匀;接种于10cm大皿中,37℃、5%co2培养箱内培养;b)第二天观察细胞状态,长至70%左右,状态良好;取2个无菌1.5ml离心管置于离心管架分别a管和b管,取650ulopti-mem 加入到两个离心管,取36μl lipofectamine 2000于a管中,分别取8ug质粒 3*flag-pparγ-egfp、6μgplp1、6μg plp2、6μg vsvg 加入b管中,室温静置5min;c)将长至80%的293t培养基弃掉,pbs清洗后换成opti-mem培养基培养;d)5min后,将a、b两管混合,室温静置20min;e)将a、b混合液转移至293t细胞的培养液中,混匀,于37℃、5% co2细胞培养箱中培养;f)培养6 h后弃去含有a、b混和物的培养基, pbs清洗,加入含10%灭活血清的dmem细胞培养基6 ml,于37℃、5% co2培养箱内继续培养48 h;g)收集上清,1000 rpm离心5min,弃去细胞碎片,上清液用0.45 μm pvdf滤器过滤至5 ml圆底离心管中;(4)利用ptg8000浓缩病毒液的方法浓缩病毒液;(5)利用电转仪通过电转的方式将浓缩病毒转入raw264.7细胞;具体步骤如下:a)将长至70%-80%的两皿raw264.7细胞拿到安全柜中,吹打下细胞,收集细胞沉淀,230g,3min离心;b)弃上清,pbs清洗一遍,分装为两管,230g,3min离心,弃上清;c)电转缓冲液为pbs和灭活的胎牛血清,用1ml血清/pbs重悬,计数,取5
×
106 个细胞(不超过200μ)于10 μl病毒浓缩液中吹打均匀,使用电极杯配套吸管加入到2mmbtx电极杯中,设置电压为200v,脉冲长度为8毫秒,脉冲数量为1,脉冲场强为1000v/cm,总样本体积为200
µ
l,温度为室温;d)将电转后的细胞悬液吸出加入含有1ml灭活的胎牛血清的六孔板中,37℃、5% co2培养箱内中培养;e)待细胞个数增长较多时,加入puro2μg/ml(一周左右)进行筛选阳性细胞。扩大
培养后,进行验证冻存;(6)稳定表达细胞的筛选;(7)细胞扩增;(8)获得稳定高表达目的基因的raw264.7细胞株。
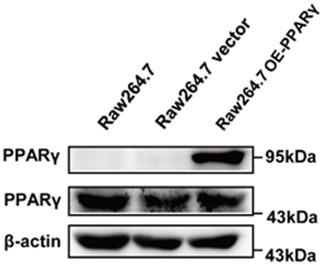
18.此外,通过该实施例在raw264.7细胞中构建的pparγ过表达稳定细胞株进行鉴定,利用western blot方法检测该细胞株中pparγ表达情况,如图1,2中,pparγ的表达明显增强,flag也明显表达。
19.将本发明实施例构建成的高表达pparγ的raw264.7细胞株进行western blot检测蛋白变化,具体包括以下步骤:(1)将细胞株分为三组进行实验,分别为:raw264.7组、raw264.7vector组、raw264.7oe-pparγ组;(2)提取蛋白;(3)将各实验组细胞收集230g,3min离心,弃上清;用预冷pbs洗3遍,瞬离吸干pbs;加入100μl ebc裂解液(4种蛋白酶抑制剂pmsf,naf,na2vo3,pi按照1:100的比例添加)置于冰上30min,每隔10min弹一弹,4℃,12000g,离心30min;小心吸取上清,转移至干净1 .5ml离心管,bca法测定蛋白浓度后,加入5xsdsloadingbuffer制样,沸水煮十分钟;(4)制胶;根据pparγ融合蛋白分子大小,分别制作分离胶浓度8%sds-page聚丙烯酰胺凝胶;(5)按照聚丙烯酰胺凝胶附带说明书进行配制:8% sds-page:水 6.9 ml,30% 丙烯酰胺溶液4ml,1.5 mol/l tris (ph8.8) 3.8ml ,10% sds 0.15ml ,10% 过硫酸氨0.15ml ,temed 0.009ml;(6)按说明书比例配制好分离胶溶液,迅速混匀后小心加入胶槽,加入无水酒精封闭压平液面,静止至少半小时,待分离胶凝固倒出残余酒精,用滤纸吸干或等待酒精挥发;(7)按照说明书比例配制压缩胶溶液,水4.2ml,30%的丙烯酰胺混合液1ml,1.0mol/ltris 溶液(ph6.8)0.75ml,10%的sds 0.06ml,10%过硫酸铵 0.06ml,temed 0.006ml;迅速混匀后加到分离胶上,插入十孔梳子,避免气泡的产生;静止至少半小时,待压缩胶凝固,准备上样电泳;(8)电泳;取下制好的胶槽放入垂直电泳槽中,加入1
×
甘氨酸电泳缓冲液,高度高于矮的玻片,外槽加入同样的1
×
甘氨酸电泳缓冲液;70v电压压齐,待溴酚蓝跑过压缩胶,marker开始分离,加大电压至100v,37kda跑到接近凝胶底部,停止电泳,取出胶板,用刮板刮开双层玻璃片,再用刮板沿着分离胶最上沿切去上端压缩胶,用刮板将胶块放入盛有转印缓冲液的器皿中,准备转膜;(9)转膜;拿出转膜的滤纸夹板,在转印缓冲液中浸湿;裁剪和凝胶大小一致的pvdf膜2张,在甲醇中浸泡约1min,然后用镊子夹至滤纸上,再用刮板把凝胶轻轻放于膜上,避免气泡的产生,最后用另一张滤纸盖住,把“三明治”夹板卡在电转卡槽中,凝胶在负极,膜在正极;将电转卡槽置于电泳槽中,开启电源,恒压100v,低温转90min;(10)5%脱脂奶粉封闭2小时;(11)孵抗体,4℃孵育过夜;收取一抗,用tbst漂洗8min
×
3次,加入二抗,置于摇床20转,常温2小时,tbst漂洗8min
×
3次;
(12)显影;根据ecl发光试剂盒说明,按照1:1的比例配好发光液,加于膜表面,用凝胶成像系统照相即可。
20.实验结果图1,2为western blot实验结果。3*flag-pparγ-egfp的表达效果如图1,2所示,在过表达flag-pparγ(raw264.7 oe-pparγ)的细胞中,在95kda附件出现了特异性的pparγ条带和falg条带,表明raw264.7细胞中,flag-pparγ稳定过表达成功。
21.将本发明实施例构建成的高表达pparγ的raw264.7细胞株进行qpcr检测相关炎症因子的变化,具体包括以下步骤:(1)将细胞株分为两组进行实验,分别为:raw264.7vector组、raw264.7oe-pparγ组;(2)使用全式金trozol按照说明书提取传代培养后raw264.7vector、raw264.7oe-pparγ细胞的mrna,并进行逆转录,得到cdna;(3)使用全式金实时荧光定量pcr试剂盒进行qpcr检测tnf-α,il-6,il-1β三种炎症因子的变化。
22.如图3所示,结果显示,raw24.7 oe-pparγ细胞中过表达的pparγ显著抑制了炎症因子tnf-α、il-6、il-1β的表达。****:p《0.0001。与vector组进行比较。
23.实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均可通过市售购买获得的常规产品。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1