一组用于东亚群体生物地理来源鉴识研究的SNP位点及方法
一组用于东亚群体生物地理来源鉴识研究的snp位点及方法
技术领域
1.本发明涉及基因鉴别技术领域,特别是涉及一组用于东亚群体生物地理来源鉴识研究的snp位点及方法。
背景技术:
2.始祖信息标记是指在群体中表现出较高等位基因频率差异的遗传标记,它可以解析未知个体的生物地理来源,也可用于识别群体中潜在的亚结构。前者在法医学研究中可为司法侦查工作提供指向性线索;后者在全基因组关联分析中可控制群体分层因素,进而可避免假阳性或者假阴性的结果。目前,法医学家通常关注的是主要洲际群体的鉴别研究。迄今为止,已有多组用于不同洲际群体法医学始祖分析的始祖信息标记被报道。然而,对于处于同一个洲际的群体或者那些位于主要洲际群体间的群体,对其法医学始祖解析的研究相对较少。
3.对于未知个体的生物地理来源的解析,法医学家通常采用主成分分析或者群体遗传结构分析的方法。前者是根据所有位点信息对所有样本进行降维分析,将变量信息转变为若干个重要的主成分,每个样本在不同主成分上都有一个特定的位置,然后根据样本在不同主成分上的分布情况,推测个体可能的生物地理来源。后者是基于贝叶斯原理,估计个体的始祖成分比例,然后通过和参考群体进行比较,根据始祖成分分布情况,确定个体的始祖来源。然而,这两种方法对于具有混合历史的个体,可能无法得出更为准确的预测结果。
4.单核苷酸多态性(single nucleotide polymorphism,snp)是由于基因组中单个核苷酸变异所形成的一种序列多态性,它具有基因组中分布广泛,突变率低的优势,在法医学研究中具有较高的应用价值。此外,既往的研究发现一些snp在不同群体中表现出较高的等位基因频率分布差异,可作为始祖信息标记用于解析不同群体的生物地理来源。
技术实现要素:
5.本发明的目的是提供一组用于东亚群体生物地理来源鉴别的snp位点及方法,以解决上述现有技术存在的问题,利用这些位点可以对北京汉族、南方汉族、傣族、日本人和越南京族进行鉴别。
6.为实现上述目的,本发明提供了如下方案:
7.本发明提供一组用于东亚群体生物地理来源鉴识研究的全基因组的snp位点的检测试剂在制备鉴别东亚群体生物地理来源的试剂盒中的应用,所述snp位点包括下表所示的位点:
8.9.10.11.12.13.14.15.16.[0017][0018]
优选的是,所述东亚群体生物地理来源包括北京汉族、南方汉族、傣族、日本人和越南京族。
[0019]
本发明还提供一种解析东亚群体生物地理来源的方法,包括筛选所述的一组用于东亚群体生物地理来源鉴识研究的全基因组的snp位点的步骤。
[0020]
优选的是,具体包括以下步骤:
[0021]
(1)基于国际千人基因组中的东亚群体的全基因组数据,采用plink软件系统初筛在东亚群体中具有相对高分化的snp位点;
[0022]
(2)采用xgboot机器学习算法对步骤(1)中初筛得到的snp位点,基于最优子集的方法进行再次筛选,最终确定能用于解析东亚群体生物地理来源的snp位点。
[0023]
优选的是,步骤(1)中,所述初筛在东亚群体中具有相对高分化的snp位点的原则包括:
[0024]
(1)日本人和非日本群体的固定系数大于0.2;
[0025]
(2)北京汉族和南方汉族的固定系数大于0.06;
[0026]
(3)傣族和越南京族人的固定系数大于0.06;
[0027]
(4)汉族群体和傣族、京族人的固定系数大于0.06;
[0028]
(5)选择的snp位点在每个群体中的最小等位基因频率大于0.01;
[0029]
(6)选择的snp位点在每个群体中均符合hwe,p值大于0.0001;
[0030]
(7)选择的snp位点的配对r2小于0.6。
[0031]
优选的是,在步骤(1)和步骤(2)之间还包括:对步骤(1)中初筛得到的snp位点采用主成分分析的方法评估其对于东亚群体的解析效能。
[0032]
优选的是,还包括:采用步骤(2)中再次筛选得到的snp位点构建预测模型,并对东亚群体进行检测分析。
[0033]
本发明还提供所述的一组用于东亚群体生物地理来源鉴识研究的全基因组的snp位点在法医学以及群体遗传学研究中的应用。
[0034]
本发明公开了以下技术效果:
[0035]
本发明提供一组在东亚群体中具有较高遗传分化的snp位点,相比既往针对不同洲际的群体,本发明中的位点可很好的用于解析东亚群体的生物地理来源,这可为法医学以及群体遗传学研究提供更有价值的信息。
[0036]
本发明提供一种基于snp位点解析东亚群体生物地理来源的方法,相比既往采用的主成分分析以及群体遗传结构分析的方法,本发明中的方法操作简便、快捷,结果准确且便于解读。
附图说明
[0037]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获
得其他的附图。
[0038]
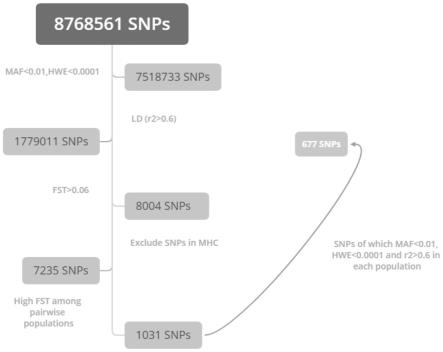
图1为snp位点筛选的流程图;
[0039]
图2为基于全基因组水平的snp位点(a)和筛选的677个snp位点(b)进行东亚群体的主成分分析;cdx:傣族;chb:北京汉族;chs:南方汉族;jpt:日本人;khv:越南京族;
[0040]
图3为不同位点获取的预测结果和实际样本结果的混淆矩阵图;a:677个snp位点的混淆矩阵图;b:258个snp位点的混淆矩阵图。
具体实施方式
[0041]
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
[0042]
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
[0043]
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
[0044]
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本技术说明书和实施例仅是示例性的。
[0045]
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
[0046]
实施例1一种解析东亚群体生物地理来源的方法
[0047]
本发明中使用到的软件主要有plink、ymodel以及r软件。涉及用于5个东亚群体生物地理来源鉴识研究的snp位点的筛选:北京汉族、南方汉族、傣族、日本人和越南京族。
[0048]
首先,5个东亚群体相对高分化的snp位点的初筛。从国际千人基因组中下载东亚群体的全基因组数据。采用plink软件,输入代码如下
‘
plink
‑‑
bfile all
‑‑
hwe 0.0001
‑‑
maf 0.01
‑‑
make-bed
‑‑
out new’,基于所有东亚个体,剔除那些hwe的p值小于0.0001和最小等位基因频率小于0.01的snp位点;接下来,采用
‘
plink
‑‑
bfile new
‑‑
indep-pairwise 50 5 0.6’,保留那些配对r2值小于0.6的snp位点;采用
‘
plink
‑‑
bfile new3
‑‑
within pop.txt
‑‑
fst’计算每个位点在东亚群体中的固定系数,选择固定系数》0.06的snp位点;剔除那些位于mhc区域的位点;对接下来的位点进行再次筛选,甄选那些在配对群体间具有较高遗传分化的位点,具体原则如下:
[0049]
1)日本人和非日本群体的固定系数大于0.2;
[0050]
2)北京汉族和南方汉族的固定系数大于0.06;
[0051]
3)傣族和越南京族人的固定系数大于0.06;
[0052]
4)汉族群体和傣族、越南京族人的固定系数大于0.06。
[0053]
最后,我们采用
‘
plink
‑‑
bfile all
‑‑
hwe 0.0001
‑‑
maf 0.01
‑‑
within pop.txt
‑‑
make-bed
‑‑
out new’和
‘
plink
‑‑
bfile new
‑‑
within pop.txt
‑‑
indep-pairwise 50 5 0.6’,对上述位点再次过滤,剔除在每个群体中hwe的p值小于0.0001和最小等位基因频率小于0.01的snp位点以及配对的r2值大于0.6的snp位点。最终,本发明保留677个snp位点。上述所述的位点筛选的流程图如图1所示。
[0054]
接下来,本发明采用目前常用的主成分分析的方法评估677个snp位点对于东亚群体的解析效能。具体操作如下:
[0055]
采用plink软件对5个东亚群体进行主成分分析,代码为
‘
plink
‑‑
bfile new1
‑‑
pca5
‑‑
out new1’;根据获得的结果,采用r软件绘制所有个体在前两个主成分上的散点图;此外,我们也对所有位点进行主成分分析。不同位点主成分分析的结果见图2。结果表明:本发明选择的677个snp位点可达到和全基因组位点相似的群体鉴别水平。
[0056]
对677个snp位点的再次筛选:本发明采用xgboot这一机器学习算法,对677个snp位点,采用最优子集的方法进行再次甄选,最终确定258个snp位点。采用677和258个snp位点分别构建预测模型,并对东亚群体进行检测分析,获得的预测结果和实际样本结果的混淆矩阵,如图3所示。不同位点构建模型的预测结果和实际结果的准确度和kappa系数,见表1。结果表明:最终确定的258个snp位点,相比选择的677个snp位点,拥有相似的性能解析这5个东亚群体的生物地理来源。
[0057]
表1选择的677个和258个snp位点对东亚群体鉴别性能的比较
[0058][0059]
以上所述的实施例仅是对本发明的优选方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1