沙库必曲中间体、其制备方法及其应用与流程
1.本发明属于医药领域,涉及医药中间体,具体涉及沙库必曲中间体、其制备方法及其应用。
背景技术:
2.哺乳动物内源性心钠肽(anp)也称为心纳素(anf),具有利尿、排钠和血管舒张功能。天然anf肽经代谢引起失活,特别是通过被认为相当于nep的降解酶失活,该酶还会引起脑啡肽的代谢失活。
3.沙库必曲(ahu-377)是由诺华公司研发的一种用于抗心衰的药物lcz696(cas:936623-90-4)的主要成分之一。该抗心衰药物由缬沙坦和沙库必曲(ahu-377)通过非共价键结合而成的超分子络合物(复合物),具有血管紧张素受体阻断和中性内肽酶抑制双重作用,降低心血管疾病的危险,主要用于治疗心脏衰竭,也可以用于高血压。
4.沙库必曲(ahu-377)通常经过关键中间体n-boc氨基醇[式(10-a)]进行制备,该中间体化学名称为:n-[(1r)-2-[1,1'-联苯]-4-基-1-(羟基甲基)乙基]氨基甲酸叔丁酯(cas:1426129-50-1);其结构式为:
[0005][0006]
现有技术中关于沙库必曲中间体n-boc氨基醇合成方法的专利文献有很多,如文献j.med.chem.1995,38,1689-1700,报道了一种以d-酪氨酸为原料,制备沙库必曲中间体的方法。合成路线如下:
[0007][0008]
该方法使用的d-酪氨酸为非天然氨基酸,价格昂贵;反应过程还使用到昂贵的三氟甲磺酸酐试剂,并且该试剂很活泼,腐蚀性强,对生产设备和操作要求高,不利于工业应用。
[0009]
专利wo2014032627和专利cn 105026361公开了n-boc氨基醇的制备方法,合成路线如下所示:
[0010][0011]
该方法主要存在的问题有:使用到三苯基膦,反应后生成大量的三苯氧膦化合物,导致分离纯化困难,固体废料多;还使用了偶氮二甲酸酯类化合物,这些化合物对光、热和震动敏感,加热过程存在潜在的危险,这些问题将会导致整个生产成本的提高。
[0012]
类似的,专利cn 105985225公开了一种沙库必曲中间体的制备方法,具体如下:
[0013][0014]
该方法与专利wo2014/032627和专利cn 105026361公开的方法类似,主要改变在于使用羟基保护剂代替了环氧氯丙烷,反应过程基本类似,并且相同反应类型都采用了基本一致的试剂,最后步骤,由于羟基使用苄基进行保护,还需要额外的钯催化氢解脱去保护基。虽然该专利报道称制备n-boc氨基醇的产率得到了提高,但是考虑试剂成本,最后增加贵金属催化氢解脱除苄基的步骤,成本上会大幅提高。
[0015]
专利wo 2013/026773和cn 103764624公开了一种通过对苯基苯甲醛为原料制备沙库必曲中间体氨基醇的方法,其合成路线如下:
[0016][0017]
该工艺中包括催化氢化步骤,其缺点是使用了贵金属rh和pd,用量较大,导致生产成本高。
[0018]
除了上述例举的一些化学合成法可以制备n-boc氨基醇中间体外,还可以通过酶方法制备该中间体。如专利cn 105884656公开了通过酶催化的不对称还原氨化制备该中间体的方法,具体如下:
[0019][0020]
在该方法中,以苄基溴化镁为原料,与草酰氯单甲酯反应生成酮酸酯;接着在溴代试剂作用下,苯环4-位发生溴化;使用铜催化与苯硼酸进行偶联,得到联苯酮酸酯;在葡萄糖、nadp
+
和还原酶cgkr2与gdh体系中,对酮酸酯进行催化不对称还原胺化,获得手性氨基酸甲酯;经过boc保护氨基后,在硼氢化钠和路易斯酸作用下,将羧酸甲酯还原为醇,得到关键中间体n-boc氨基醇。
[0021]
该方法存在合成路线较长的问题,其次酰氯、溴代试剂使用上不方便,铜催化偶联和不对称还原胺化步骤中,需要使用到较多的金属铜、还原酶;此外,该专利中并未指出经过还原胺化后,产物的对映体过量情况。
[0022]
综上所述,现有报道的制备工艺中,沙库必曲关键手性中间体n-boc氨基醇(10-a)的制备,一方面受到原料、反应试剂、后处理工艺等方面的限制,另外一方面合成路线长、非对映异构体比例低、环境不友好等问题,导致了生产成本高,操作繁琐,不利于工业化。因此,开发出关键手性中间体n-boc氨基醇(10-a)更简便、经济和便于工业化的生产路线,具有重要意义。
[0023]
相对于目前已报道路线本技术路线具有收率高,操作简便可实现连续化操作便于工艺放大生产,所用原料价格低廉易得,成本低等。
[0024]
相对于目前已报道路线本技术路线具有收率高,操作简便可实现连续化操作便于工艺放大生产,所用原料价格低廉易得,成本低等。具体地,
[0025]
1、本技术工艺路线所用的辅料如氢氧化钠,盐酸,甲磺酰氯等均为工业化大生产常用物料,价格便宜易得,经济型极好生产成本低廉。
[0026]
2、本技术工艺路线中多个中间体无需分离纯化可进行连续化投料生产,可有效降低工业生产的能耗及人工成本,如实施例1减压回收溶剂后可直接进行实施例2,实施例1步骤2中分出的有机相可直接进行成盐操作得到的产物进入水相直接进行实施例3,实施例7步骤6中包含三步反应以甲苯为溶剂可实现一锅法操作,同时完成产物(10-a)的纯化。
技术实现要素:
[0027]
本发明涉及沙库必曲中间体的制备工艺,通过本发明所公开方法的实施,可以高效制备关键中间体n-boc氨基醇[式(10)和式(10-a)]。所述的中间体可用于中性内肽酶(nep)抑制剂或其前药,尤其地,包含γ-氨基-δ-联苯-α-甲基烷酸或酯的骨架的nep抑制剂,如沙库必曲等。该工艺路线原料便宜、操作简便、对设备无特殊要求、生产成本低,适用
于工业化生产,具有极大的应用潜力和商业化价值。并且本发还提供了制备化合物(10-a)的中间体。
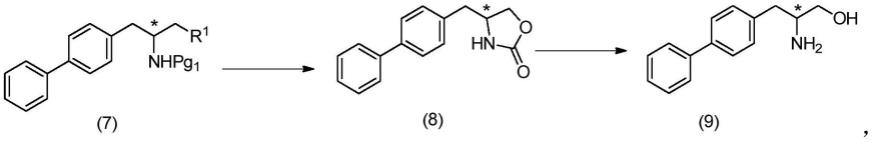
[0028]
具体地,一方面,本发明提供一种化合物(10)的制备工艺,其包括以下步骤:
[0029][0030]
步骤a:化合物(6)与化合物(m)在催化剂作用下反应得到化合物(7),其中,
[0031]“*”表示化合物(6)和化合物(7)均为r构型或s构型;优选地,“*”表示化合物(6)和化合物(7)均为r构型;
[0032]
r1为f、cl、br、i或-or3,r3表示c
1-6
烷基或c
1-6
杂烷基;
[0033]
r2为其中x为cl、br或i;
[0034]
pg1为氨基保护基;
[0035]
化合物(10)为结构:其中,“*”表示化合物(10)为r构型或s构型,优选地,“*”表示化合物(10)为r构型。
[0036]
在一些实施方案中,本发明所述的工艺,步骤a中,
[0037]
所述催化剂为亚铜盐,优选地为cui、cubr、cucl或cucn;所述氨基保护基为苄氧羰基、叔丁氧羰基、甲氧羰基、乙氧羰基、异丙基氧羰基、异丁基氧羰基、笏甲氧基羰基、烯丙氧羰基或三甲基硅乙氧羰基;或
[0038]
所述反应是在干燥无氧条件下进行的,优选地,所述反应是在干燥无氧条件下通入氮气保护进行的;或
[0039]
所述反应的反应温度为-60℃~0℃,优选反应温度为-45℃~0℃,更优选反应温度为-30℃~-10℃,最优选反应温度为-20℃~-15℃;或
[0040]
所述反应的反应时间为0.5h~3h;优选反应时间为0.8h~2h;更优选反应时间为1h~1.5h;最优选反应时间为1h。
[0041]
在一些实施方案中,本发明所述的工艺,步骤a中,所述反应的反应溶剂为第一溶剂,所述第一溶剂为有机非质子溶剂;优选地,所述有机非质子溶剂为四氢呋喃、2-甲基四氢呋喃、甲苯、环戊烷甲醚、二氯甲烷、甲基叔丁基醚或乙醚,或其任意组合。
[0042]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)与化合物(m)的反应摩尔当量比为1:0.7~2,优选1:1~2,更优选1:1~1.5,最优选1:1。
[0043]
在一些实施方案中,本发明所述的工艺,还包括以下步骤:
[0044][0045]
其中,“*”表示化合物(7)、化合物(8)和化合物(9)均为r构型或s构型,优选地,“*”表示化合物(7)、化合物(8)和化合物(9)均为r构型;
[0046]
步骤b:化合物(7)在第二溶剂中和第一加热条件下反应得到化合物(8);
[0047]
其中优选地,所述反应时间为0.3h~2h,更选地,所述反应时间为0.3h~1h,最优选地,所述反应时间为0.5h;
[0048]
步骤c:化合物(8)在第三溶剂中和第二加热条件下与碱进行水解反应得到化合物(9);其中,
[0049]
优选地,所述碱为氢氧化钠、氢氧化钾、氢氧化锂、叔丁醇钾、叔丁醇钠、碳酸钠、碳酸钾或甲醇钠或其任意组合;
[0050]
优选地,所述化合物(8)和碱的摩尔当量比为1:(0.9~5),更优选地,所述化合物(8)和碱的摩尔当量比为1:(1~4),更进一步优选地,所述化合物(8)和碱的摩尔当量比为1:(2~4),最优选地,所述化合物(8)和碱的摩尔当量比为1:3.75;
[0051]
优选地,所述反应时间为1h~5h,更选地,所述反应时间为2h~4h,最优选地,所述反应时间为3h。
[0052]
在一些实施方案中,本发明所述的工艺,步骤b和步骤c中,所述第一加热条件和第二加热条件下的加热温度各自独立地为80℃~150℃,优选地为100℃~150℃,进一步优选地为110℃~130℃,最优选地为120℃;优选地,所述第一加热条件和第二加热条件下的加热温度为相同温度;或,
[0053]
所述第二溶剂和第三溶剂各自独立地为正丁醇、苯、甲苯、乙二醇二甲醚、n,n-二甲基甲酰胺、n.n-二甲基乙酰胺;优选地,所述第二溶剂和第三溶剂为相同溶剂。
[0054]
在一些实施方案中,本发明所述的工艺,所述步骤b反应结束后不经后处理直接加入碱进行步骤c的反应。
[0055]
在一些实施方案中,本发明所述的工艺,还包括以下步骤:
[0056][0057]
其中,“*”表示化合物(9)和化合物(10)均为r构型或s构型,优选地,“*”表示化合物(9)和化合物(10)均为r构型;
[0058]
步骤d:化合物(9)通过氨基保护反应得到化合物(10)。
[0059]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)由以下步骤制备而成:
[0060][0061]
其中,化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)上的“*”表示化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)均为s构型,化合物(6)上的“*”表示化合物(6)为r构型;或,化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)上的“*”表示化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)均为r构型,化合物(6)上的“*”表示化合物(6)为s构型;
[0062]
pg2为羟基保护基,优选为甲磺酰基、三氟甲磺酰基、对甲苯磺酰基或硝基磺酰基;r1和pg1具有本发明所述的定义;
[0063]
步骤a’:化合物(2)与羟基保护试剂通过羟基保护反应得到化合物(3);优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:(1~3),更优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:(1~2),更进一步优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:(1.2~1.5),最优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:1.3;
[0064]
步骤b’:化合物(3)与酸hy反应得到化合物(4a);优选地,所述酸hy为盐酸、氢溴酸、甲酸、氢碘酸、对甲苯磺酸或三氟甲磺酸;
[0065]
步骤c’:化合物(4a)在碱作用下得到化合物(4);优选地,所述碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸氢钠、碳酸氢钾或碳酸钾;
[0066]
步骤d’:化合物(4)与氨基保护试剂通过氨基保护反应得到化合物(5);优选地,所述化合物(4)与氨基保护试剂的摩尔当量比为1:1~1.5;
[0067]
步骤e’:化合物(5)在碱性试剂作用下在第四溶剂中反应得到化合物(6);优选地,所述碱性试剂为氢化钠、甲醇钠、氢氧化钠、氢氧化钾、叔丁醇钾、叔丁醇钠、碳酸钾、碳酸钠或乙醇钠;优选地,所述化合物(5)与碱性试剂的摩尔当量比为1:(1~3),更优选为1:(1.5~2.5),最优选为1:2.2;优选地,所述第四溶剂为四氢呋喃、n,n-二甲基甲酰胺;优选地,所述反应温度为-10℃~50℃,进一步优选地,所述反应温度为-10℃~15℃,更进一步优选地,所述反应温度为-5℃~10℃,最优选地,所述反应温度为0℃~10℃。
[0068]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)由以下步骤制备而成:
[0069][0070]
其中,化合物(4-1-a)和化合物(5-1)上的“*”表示化合物(4-1-a)和化合物(5-1)均为s构型,化合物(6)上的“*”表示化合物(6)为r构型;或,化合物(4-1-a)和化合物(5-1)
上的“*”表示化合物(4-1-a)和化合物(5-1)均为r构型,化合物(6)上的“*”表示化合物(6)为s构型;
[0071]
步骤a”:化合物(4-1-a)在碱作用下与氨基保护试剂进行氨基保护反应得到化合物(5-1);优选地,所述化合物(4-1-a)与氨基保护试剂的摩尔当量比为1:(0.7~2);优选地,所述碱为氢氧化钠、氢氧化钾、碳酸钠或碳酸钾;
[0072]
步骤b”:化合物(5-1)在三苯基膦和偶氮二甲酸乙酯作用下和氮气保护下低温反应得到化合物(6),其中,所述低温为-40℃~0℃,优选为-30℃~-10℃,更优选为-20℃~-10℃。
[0073]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)由以下步骤制备而成:
[0074]
其中,
[0075]
化合物(4a)上的“*”表示化合物(4a)为s构型,化合物(5-2)和化合物(6)上的“*”表示化合物(5-2)和化合物(6)均为r构型;或,化合物(4a)上的“*”表示化合物(4a)和r构型,化合物(5-2)和化合物(6)上的“*”表示化合物(5-2)和化合物(6)均为s构型;
[0076]
步骤a
”’
:化合物(4a)在碱作用下反应得到化合物(5-2);优选地,所述碱为氢氧化钠、氢氧化钾、碳酸钠或碳酸钾;优选地,所述化合物(4a)与碱的摩尔当量比为1:(1~3),更优选为1:(1.2~2.5),最优选为1:1.5;优选地,所述反应温度为10℃~100℃,更优选地,所述反应温度为30℃~80℃,更进一步优选地,所述反应温度为40℃~60℃,最优选地,所述反应温度为50℃;
[0077]
步骤b
”’
:化合物(5-2)继续与氨基保护试剂进行氨基保护反应得到化合物(6);优选地,所述化合物(5-2)与氨基保护试剂的摩尔当量比为1:(0.7~2);优选地,所述反应为低温反应,所述低温为为-5℃~15℃,更优选所述低温为0℃~10℃。
[0078]
另一方面,本发明提供一种化合物,其具有如下式(7a)或(7b)结构:
[0079][0080]
其中,r
1a
为f、cl、br、i或-or3,r3表示c
1-6
烷基或含杂原子取代的c
1-6
烷基;
[0081]
pg
1a
为氨基保护基,优选为苄氧羰基、叔丁氧羰基、甲氧羰基、乙氧羰基、异丙基氧羰基、异丁基氧羰基、笏甲氧基羰基、烯丙氧羰基或三甲基硅乙氧羰基;
[0082]
且当r
1a
为br或i时,pg
1a
不为叔丁氧羰基。
[0083]
在一些实施方案中,本发明化合物(7a)或化合物(7b)具有如下其中之一结构:
[0084][0085]
另一方面,本发明提供一种化合物,其具有如下式(6a)或(6b)结构:
[0086][0087]
其中,x为f、cl、br或i;
[0088]
pg1为氨基保护基,优选为苄氧羰基、叔丁氧羰基、甲氧羰基、乙氧羰基、异丙基氧羰基、异丁基氧羰基、笏甲氧基羰基、烯丙氧羰基或三甲基硅乙氧羰基。
[0089]
在一些实施方案中,本发明化合物(6a)或化合物(6b)具有如下其中之一结构:
[0090][0091]
另一方面,本发明提供一种化合物,其具有如下式(8-a)或(8-b)结构:
[0092][0093]
另一方面,本发明提供本发明所述的工艺或本发明所述的化合物在制备沙库必曲中的应用。
[0094]
本发明的详细说明书
[0095]
术语定义
[0096]
现在详细描述本发明的某些实施方案,其实例由随附的结构式和化学式说明。本发明意图涵盖所有的替代、修改和等同技术方案,它们均包括在如权利要求定义的本发明范围内。本领域技术人员应认识到,许多与本文所述类似或等同的方法和材料能够用于实践本发明。本发明绝不限于本文所述的方法和材料。在所结合的文献、专利和类似材料的一篇或多篇与本技术不同或相矛盾的情况下(包括但不限于所定义的术语、术语应用、所描述
的技术,等等),以本技术为准。
[0097]
除非另外说明,本发明所使用的所有科技术语具有与本发明所属领域技术人员的通常理解相同的含义。本发明涉及的所有专利和公开出版物通过引用方式整体并入本发明。
[0098]
在本发明中所采用的描述方式“各
…
独立地为”与
“…
各自独立地为”和
“…
独立地为”可以互换,均应做广义理解,其既可以是指在不同基团中,相同符号之间所表达的具体选项之间互相不影响,也可以表示在相同的基团中,相同符号之间所表达的具体选项之间互相不影响。
[0099]
本发明中“c
q1-q2”表示所描述的基团的碳原子个数,例如,c
1-6
烷基表示含有1-6个碳原子的烷基。
[0100]
术语“烷基”表示含有1至20个碳原子,饱和的直链或支链一价烃基基团。在另一实施方案中,烷基基团含有1-6个碳原子;在又一实施方案中,烷基基团含有1-4个碳原子;还在一实施方案中,烷基基团含有1-3个碳原子。烷基基团的实例包含,但并不限于,甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基(s-bu、-ch(ch3)ch2ch3),叔丁基、正戊基、2-戊基,等。
[0101]
术语“杂烷基”表示烷基基团中一个碳原子被杂原子所取代,其中杂原子为o、s、n、si。在一实施方案中,杂烷基基团含有1-6个碳原子;在又一实施方案中,杂烷基基团含有1-4个碳原子;还在一实施方案中,杂烷基基团含有1-3个碳原子。杂烷基基团的实例包含,但并不限于,-ch2och3、-ch2och
2 ch3、-ch2sch3、-ch2sih2ch3、-ch2nhch3,等。
[0102]
术语“卤素”表示f(氟)、cl(氯)、br(溴)或i(碘)。
[0103]
术语“氨基保护基”是指一个取代基与氨基基团相连来阻断或保护化合物中氨基的功能性,氨基保护基的实例包括但不限于苄氧羰基(-cbz)、叔丁氧羰基(-boc)、甲氧羰基(-coome或-co2me)、乙氧羰基(-cooet或-co2et)、异丁基氧羰基(-cooibu或-co
2i
bu)、异丙基氧羰基(-cooipr或-co
2i
pr)、笏甲氧基羰基(fmoc)、烯丙氧羰基(alloc)、三甲基硅乙氧羰基(teoc)。
[0104]
术语“羟基基保护基”,是指羟基的取代基用来阻断或保护羟基的功能性,羟基保护基的实例包括但不限于,甲磺酰基-(ms)、三氟甲磺酰基(-tf)、对甲苯磺酰基(-ts)或硝基磺酰基(-ns)
[0105]
术语“第一”或“第二”或“第三”不表示先后顺序,仅是为了区分第一或第二或第三后面的修饰词,如第一溶剂、第二溶剂或第三溶剂分别表示对应反应中所用的溶剂;同样,第一加热条件和第二加热条件也是分别表示对应反应中的加热条件。
[0106]
本发明的详细描述。
[0107]
通过本发明所公开方法的实施,可以高效制备关键中间体n-boc氨基醇[式(10)或式(10-a)]。所述的中间体可用于中性内肽酶(nep)抑制剂或其前药,尤其地,包含γ-氨基-δ-联苯-α-甲基烷酸或酯的骨架的nep抑制剂,如沙库必曲等。并且本发还提供了制备化合物(10)或化合物(10-a)的中间体。
[0108]
具体地,一方面,本发明提供一种化合物(10)的制备工艺,其包括以下步骤:
[0109][0110]
其中,r1、r2、pg1和化合物(10)各自具有如本发明所述的定义;
[0111]
步骤a:化合物(6)与化合物(m)在催化剂作用下反应得到化合物(7),其中,
[0112]“*”表示化合物(6)和化合物(7)均为r构型或s构型。
[0113]
在一些优选的实施方案中,步骤a中,“*”表示化合物(6)和化合物(7)均为r构型,则步骤a表示为:
[0114][0115]
步骤[a]:化合物(6a’)与化合物(m)在催化剂作用下反应得到化合物(7a’),其中r1、r2和pg1各自具有如本发明所述的定义。
[0116]
在一些实施方案中,r1为f、cl、br、i或-or3,r3表示c
1-6
烷基或c
1-6
杂烷基;
[0117]
在一些实施方案中,r2为其中x为cl、br或i。
[0118]
在一些实施方案中,pg1为氨基保护基。
[0119]
在一些实施方案中,化合物(10)为结构:其中,“*”表示化合物(10)为r构型或s构型。
[0120]
在一些优选的实施方案中,化合物(10)为结构:其中,“*”表示化合物(10)为r构型,即化合物(10)为化合物(10-a):
[0121]
在一些实施方案中,本发明所述的工艺,步骤a中,所述催化剂为亚铜盐,优选地为cui、cubr、cucl或cucn。
[0122]
在一些实施方案中,本发明所述的工艺,步骤a中,所述氨基保护基为苄氧羰基、叔
丁氧羰基、甲氧羰基、乙氧羰基、笏甲氧基羰基、烯丙氧羰基或三甲基硅乙氧羰基。
[0123]
在一些实施方案中,本发明所述的工艺,步骤a中,所述反应是在干燥无氧条件下进行的;在一些优选的实施方案中,本发明所述的工艺,步骤a中,所述反应是在干燥无氧条件下通入氮气保护进行的。
[0124]
在一些实施方案中,本发明所述的工艺,步骤a中,所述反应的反应温度为-60℃~0℃;在一些优选的实施方案中,本发明所述的工艺,步骤a中,所述反应温度为-45℃~0℃,在一些更优选的实施方案中,本发明所述的工艺,步骤a中,所述反应温度为-30℃~-10℃;在一些最优选的实施方案中,本发明所述的工艺,步骤a中,所述反应温度为-20℃~-15℃。
[0125]
在一些实施方案中,本发明所述的工艺,步骤a中,所述反应的反应时间为0.5h~3h;在一些优选的实施方案中,本发明所述的工艺,步骤a中,所述反应时间为0.8h~2h;在一些更优选的实施方案中,本发明所述的工艺,步骤a中,所述反应时间为1h~1.5h;在一些最优选的实施方案中,本发明所述的工艺,步骤a中,所述反应时间为1h。
[0126]
在一些实施方案中,本发明所述的工艺,步骤a中,所述反应的反应溶剂为第一溶剂,所述第一溶剂为有机非质子溶剂;优选地,所述有机非质子溶剂为四氢呋喃、2-甲基四氢呋喃、甲苯、环戊烷甲醚、二氯甲烷、甲基叔丁基醚或乙醚,或其任意组合。
[0127]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)与化合物(m)的反应摩尔当量比为1:0.7~2;在一些优选的实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)与化合物(m)的反应摩尔当量比为1:1~2;在一些更优选的实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)与化合物(m)的反应摩尔当量比为1:1~1.5;在一些更优选的实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)与化合物(m)的反应摩尔当量比为1:1。
[0128]
在一些实施方案中,本发明所述的工艺,还包括以下步骤:
[0129][0130]
其中,r1、r2、pg1和化合物(10)各自具有如本发明所述的定义;
[0131]“*”表示化合物(7)、化合物(8)和化合物(9)均为r构型或s构型;优选地,“*”表示化合物(7)、化合物(8)和化合物(9)均为r构型;
[0132]
步骤b:化合物(7)在第二溶剂中和第一加热条件下反应得到化合物(8);
[0133]
其中优选地,所述反应时间为0.3h~2h,更选地,所述反应时间为0.3h~1h,最优选地,所述反应时间为0.5h;
[0134]
步骤c:化合物(8)在第三溶剂中和第二加热条件下与碱进行水解反应得到化合物(9);其中,
[0135]
优选地,所述碱为氢氧化钠、氢氧化钾、氢氧化锂、叔丁醇钾、叔丁醇钠、碳酸钠、碳酸钾或甲醇钠或其任意组合;
[0136]
优选地,所述化合物(8)和碱的摩尔当量比为1:(0.9~5),更优选地,所述化合物(8)和碱的摩尔当量比为1:(1~4),更进一步优选地,所述化合物(8)和碱的摩尔当量比为1:(2~4),最优选地,所述化合物(8)和碱的摩尔当量比为1:3.75;
[0137]
优选地,所述反应时间为1h~5h,更选地,所述反应时间为2h~4h,最优选地,所述反应时间为3h。
[0138]
在一些实施方案中,所述第一加热条件和第二加热条件下的加热温度各自独立地为80℃~150℃,优选地为100℃~150℃,进一步优选地为110℃~130℃,最优选地在一些实施方案中,本发明所述的工艺,步骤b和步骤c中,为120℃;优选地,所述第一加热条件和第二加热条件下的加热温度为相同温度。
[0139]
在一些实施方案中,本发明所述的工艺,步骤b和步骤c中,所述第二溶剂和第三溶剂各自独立地为正丁醇、苯、甲苯、乙二醇二甲醚、n,n-二甲基甲酰胺、n.n-二甲基乙酰胺;优选地,所述第二溶剂和第三溶剂为相同溶剂。
[0140]
在一些实施方案中,本发明所述的工艺,所述步骤b反应结束后不经后处理直接加入碱进行步骤c的反应。
[0141]
在一些实施方案中,本发明所述的工艺,还包括以下步骤:
[0142][0143]
其中,
ꢀ“
*”表示化合物(9)和化合物(10)均为r构型或s构型,优选地,“*”表示化合物(9)和化合物(10)均为r构型;
[0144]
步骤d:化合物(9)通过氨基保护反应得到化合物(10)。
[0145]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)由以下步骤制备而成:
[0146][0147]
其中,化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)上的“*”表示化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)均为s构型,化合物(6)上的“*”表示化合物(6)为r构型;或,化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)上的“*”表示化合物(2)、化合物(3)、化合物(4a)、化合物(4)和化合物(5)均为r构型,化合物(6)上的“*”表示化合物(6)为s构型;
[0148]
pg2为羟基保护基,优选为甲磺酰基、三氟甲磺酰基、对甲苯磺酰基或硝基磺酰基;r1和pg1具有如权利要求1所述的定义;
[0149]
步骤a’:化合物(2)与羟基保护试剂通过羟基保护反应得到化合物(3);优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:(1~3),更优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:(1~2),更进一步优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:(1.2~1.5),最优选地,所述化合物(2)与羟基保护试剂的摩尔当量比为1:
1.3;
[0150]
步骤b’:化合物(3)与酸hy反应得到化合物(4a);优选地,所述酸hy为盐酸、氢溴酸、甲酸、氢碘酸、对甲苯磺酸或三氟甲磺酸;
[0151]
步骤c’:化合物(4a)在碱作用下得到化合物(4);优选地,所述碱为氢氧化钠、氢氧化钾、碳酸钠或碳酸钾;
[0152]
步骤d’:化合物(4)与氨基保护试剂通过氨基保护反应得到化合物(5);优选地,所述化合物(4)与氨基保护试剂的摩尔当量比为1:1~1.5;
[0153]
步骤e’:化合物(5)在碱性试剂作用下在第四溶剂中反应得到化合物(6);优选地,所述碱性试剂为氢化钠、甲醇钠、氢氧化钠、氢氧化钾、叔丁醇钾、叔丁醇钠、碳酸钾、碳酸钠或乙醇钠;优选地,所述化合物(5)与碱性试剂的摩尔当量比为1:(1~3),更优选为1:(1.5~2.5),最优选为1:2.2;优选地,所述第四溶剂为四氢呋喃、n,n-二甲基甲酰胺;优选地,所述反应温度为-10℃~50℃,进一步优选地,所述反应温度为-10℃~15℃,更进一步优选地,所述反应温度为-5℃~10℃,最优选地,所述反应温度为0℃~10℃。
[0154]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)由以下步骤制备而成:
[0155][0156]
其中,化合物(4-1-a)和化合物(5-1)上的“*”表示化合物(4-1-a)和化合物(5-1)均为s构型,化合物(6)上的“*”表示化合物(6)为r构型;或,化合物(4-1-a)和化合物(5-1)上的“*”表示化合物(4-1-a)和化合物(5-1)均为r构型,化合物(6)上的“*”表示化合物(6)为s构型;
[0157]
步骤a”:化合物(4-1-a)在碱作用下与氨基保护试剂进行氨基保护反应得到化合物(5-1);优选地,所述化合物(4-1-a)与氨基保护试剂的摩尔当量比为1:(0.7~2);优选地,所述碱为氢氧化钠、氢氧化钾、碳酸钠或碳酸钾;
[0158]
步骤b”:化合物(5-1)在三苯基膦和偶氮二甲酸乙酯作用下和氮气保护下低温反应得到化合物(6),其中,所述低温为-40℃~0℃,优选为-30℃~-10℃,更优选为-20℃~-10℃。
[0159]
在一些实施方案中,本发明所述的工艺,步骤a中,所述化合物(6)由以下步骤制备而成:
[0160]
其中,
[0161]
化合物(4a)上的“*”表示化合物(4a)为s构型,化合物(5-2)和化合物(6)上的“*”表示化合物(5-2)和化合物(6)均为r构型;或,化合物(4a)上的“*”表示化合物(4a)为r构型,化合物(5-2)和化合物(6)上的“*”表示化合物(5-2)和化合物(6)均为s构型;
[0162]
步骤a
”’
:化合物(4a)在碱作用下反应得到化合物(5-2);优选地,所述碱为氢氧化钠、氢氧化钾、碳酸钠或碳酸钾;优选地,所述化合物(4a)与碱的摩尔当量比为1:(1~3),更优选为1:(1.2~2.5),最优选为1:1.5;优选地,所述反应温度为10℃~100℃,更优选地,所述反应温度为30℃~80℃,更进一步优选地,所述反应温度为40℃~60℃,最优选地,所述反应温度为50℃;
[0163]
步骤b
”’
:化合物(5-2)继续与氨基保护试剂进行氨基保护反应得到化合物(6);优选地,所述化合物(4-1-a)与氨基保护试剂的摩尔当量比为1:(0.7~2);优选地,所述反应为低温反应,所述低温为为-5℃~15℃,更优选所述低温为0℃~10℃。
[0164]
另一方面,本发明提供一种化合物,其具有如下式(7a)或(7b)结构:
[0165][0166]
其中,r
1a
为f、cl、br、i或-or3,r3表示c
1-6
烷基或含杂原子取代的c
1-6
烷基;
[0167]
pg
1a
为氨基保护基,优选为苄氧羰基、叔丁氧羰基、甲氧羰基、乙氧羰基、异丙基氧羰基、异丁基氧羰基、笏甲氧基羰基、烯丙氧羰基或三甲基硅乙氧羰基;
[0168]
且当r
1a
为br或i时,pg
1a
不为叔丁氧羰基。
[0169]
在一些实施方案中,本发明化合物(7a)或化合物(7b)具有如下其中之一结构:
[0170][0171]
另一方面,本发明提供一种化合物,其具有如下式(6a)或(6b)结构:
[0172][0173]
其中,x为f、cl、br或i;
[0174]
pg1为氨基保护基,优选为苄氧羰基、叔丁氧羰基、甲氧羰基、乙氧羰基、异丙基氧羰基、异丁基氧羰基、笏甲氧基羰基、烯丙氧羰基或三甲基硅乙氧羰基。
[0175]
在一些实施方案中,本发明化合物(6a)或化合物(6b)具有如下其中之一结构:
[0176][0177]
另一方面,本发明提供一种化合物,其具有如下式(8-a)或(8-b)结构:
[0178][0179]
另一方面,本发明提供本发明所述的工艺或本发明所述的化合物在制备沙库必曲中的应用。
具体实施方式
[0180]
一般地,本发明的化合物可以通过本发明所描述的方法制备得到。下面的反应方案和实施例用于进一步举例说明本发明的内容。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0181]
下面所描述的实施例,除非其他方面表明所有的温度定为摄氏度。本发明的室温为10℃~30℃或15℃~25℃。若无其他说明,试剂均为常规试剂,可从市场上购买得到。色谱柱是使用硅胶柱。核磁共振光谱以cdc13或dmso-d6为溶剂(报导以ppm为单位),用tms(0ppm)或氯仿(7.25ppm)作为参照标准。偶合常数j,用赫兹(hz)表示。
[0182]
下面简写词的使用贯穿本发明:
[0183][0184][0185]
实施例
[0186]
实施例1:化合物(6-a)的合成
[0187]
步骤1:化合物(2-a)的合成
[0188][0189]
取苯甲醛50g(0.47mol)加入乙醇300ml室温搅拌,取氨水(25%)50ml在温度15~20℃下滴入反应体系,滴加完毕恢复室温搅拌20min后,取(s)-环氧氯丙烷52g(0.56mol,1.2eq)在氮气保护下用50ml乙醇稀释后缓慢滴入反应体系,滴加完毕体系室温25~30℃搅拌反应12h。gc监测反应,环氧氯丙烷消失,反应结束。减压蒸馏回收乙醇得到黄色油状产品加入100ml乙醇再次蒸馏后得到浅黄色油状液体113g为粗品(2-a)。
[0190]
表一:实施例1步骤1不同工艺条件筛选
[0191][0192]
对反应进行条件筛选优化发现,在其他条件与表一上一段落一致的情况下,在滴加环氧氯丙烷过程中氮气保护可有效降低产生的杂质,提高产率。同时反应放大后与小试结果类似,反应稳定性较好。
[0193]
步骤2:化合物(4-a)的合成
[0194][0195]
取上步粗品(2-a)(113g,0.42mol,1.0eq)加入400ml二氯甲烷中,室温搅拌有少量白色不溶物,过滤,滤液加入三乙胺63.7g(0.629mol,1.5eq),降温至內温0~10℃,滴加100ml二氯甲烷稀释的甲磺酰氯62.5g(0.545mol,1.3eq),体系內温升至12℃,有白色物质析出。滴加完毕体系降温至4℃,保温搅拌1h后正相hplc监测反应,原料消失。体系加入400ml水充分搅拌30min,萃取,有机相(400ml
×
2)水洗涤两次。取120g浓盐酸用240ml水稀释后加入有机相中,室温充分搅拌3~4h,静置分层,有机相用50ml水洗涤后合并水相得到粗品(4-a)水溶液直接用于下步反应。
[0196]
表二:实施例1步骤2不同工艺条件筛选
[0197][0198]
在与表二上一段落相似的反应条件下,对于同一批次的(2-a)进行羟基保护反应,发现反应转化率与甲璜酰氯使用量相关。使用当量为1.3eq时,可以使反应转化完全。
[0199]
步骤3:化合物(5-a)的合成
[0200][0201]
步骤2的(4-a)的盐酸盐水溶液(0.374mol,1.0eq)加入2l反应瓶内,降温內温0-10℃后滴入氢氧化钠水溶液(17g氢氧化钠加入100ml水溶解,1.1eq)调节ph值为8~9之间,体系逐渐变为白色浑浊状态,滴加完毕,体系维持0~10℃。取82g(1eq)二碳酸二叔丁酯用100ml二氯甲烷稀释后滴入反应体系,体系产生气体并放热,內温升至10~15℃,滴加完毕后保温反应2h后lc-ms监测反应原料消失,反应结束。静置分层,水相加入137g二氯甲烷萃取,分出有机相139g,合并有机相,122g水洗有机相,分出132g水相。有机相干燥过滤后减压浓缩得到淡黄色油状液体静置后变为白色蜡状固体126g为粗品(5-a)。
[0202]
(5-a)的表征数据:1h nmr(600mhz,chloroform-d)δ5.09(s,1h),4.89(s,1h),3.81-3.76(m,1h),3.70(dd,j=12.4,6.1hz,1h),3.55(dq,j=10.2,5.2hz,1h),3.45(dt,j=14.2,6.2hz,1h),3.13(s,3h),1.45(s,9h).
13
c nmr(151mhz,cdcl3)δ155.92,80.16,79.85,43.58,42.36,38.38,28.22.
[0203]
表三:实施例1步骤3不同工艺条件筛选
[0204][0205]
在与表三上一段落相似的反应条件下,当二碳酸二叔丁酯的用量为1~1.5eq时,反应均可以完全转化。
[0206]
步骤4:化合物(6-a)的合成
[0207][0208]
取步骤3的126g(5-a)粗品加入500ml四氢呋喃室温搅拌溶清,降温0~10℃,取60%的氢化钠20g(0.83mol,2eq)加入反应,体系产生气体泡沫,恢复室温25~30℃反应1h后lc-ms监测反应,原料消失。减压回收四氢呋喃后加入二氯甲烷,水萃取反应,有机相干燥脱溶得到85g淡黄色油状液体。减压蒸馏纯化得51.6g无色油状液体为(6-a),gc纯度92%,前五步合计收率58.3%。
[0209]
(6-a)的表征数据:1h nmr(600mhz,chloroform-d)δ3.63(dd,j=10.0,4.1hz,1h),3.46(dd,j=11.3,5.9hz,1h),2.78
–
2.70(m,1h),2.38(d,j=4.0hz,1h),2.12(s,1h),1.46(s,9h).
13
c nmr(151mhz,cdcl3)δ160.96,81.15,44.50,37.12,30.92,27.43.
[0210]
表四:实施例1步骤4不同工艺条件筛选
[0211][0212]
在与表四上一段落相似的反应条件下,当碱选用nah,且溶剂选用thf时,反应收率最高。
[0213]
实施例2:化合物(6-a)的合成
[0214]
步骤1:化合物(5-1-a)的合成
[0215][0216]
取实施例1步骤1制备的的113g粗品2-a(0.4mol,1.0eq)加入200ml二氯甲烷为溶剂,86g浓盐酸(1.0eq)用100ml水稀释滴入反应,滴加完毕室温25~30℃充分搅拌1h后,静置分相,有机相用50ml水洗涤后合并水相,水相降温至内温0~10℃,取17g氢氧化钠用50ml水溶解缓慢滴入反应,滴加完毕取65g二碳酸二叔丁酯(0.417mol,1eq)用100ml二氯甲烷稀释缓慢滴入反应体系,体系放热并有气体产生,滴加完毕恢复室温搅拌2h后,lc-ms检测原料转化完全,静置分相,水相加入100ml二氯甲烷洗涤,合并有机相,干燥过滤减压脱溶后得到白色固体98g为粗品(5-1-a)。
[0217]
步骤2:化合物(6-a)的合成
[0218][0219]
取上步(5-1-a)粗品98g(0.376mol,1eq)加入300ml甲苯室温搅拌溶清,氮气保护下加入三苯基膦108g(0.413mol,1.1eq)降至内温-10~-20℃,取68.7g偶氮二甲酸二乙酯(0.394mol,1.05eq)缓慢滴入反应体系,体系逐渐由无色变为黄色有固体析出,滴加完毕,保温反应2h后,lc-ms监测反应,原料消失。后处理取150ml水加入反应,室温搅拌后静置分相,有机相减压脱溶后得到黄色油状液体115g,减压蒸馏纯化得到无色油状液体64g为纯品
6-a。gc纯度93%;(2-a)来源于实施例1,从实施例1开始计算四步反应合计收率73%。
[0220]
实施例3:化合物(6-a)的合成
[0221][0222]
实施例1步骤2中所得(4-a)盐酸盐水溶液(0.356mol,1eq)降温至5~10℃,取15g氢氧化钠(0.392mol,1.1eq)用50ml水稀释后缓慢滴入反应体系,滴加完毕体系补加甲苯200ml加热升温至50℃反应1~2h后,lc-ms监测反应原料转化完毕。体系降温至0~10℃,取二碳酸二叔丁酯71.5g(0.327mol,1eq)滴入反应体系,滴加完毕恢复室温反应2~3h后lc-ms显示原料消失有目标产物生成。静置分层,水相用100ml甲苯洗涤后合并有机相,减压脱溶后得到黄色油状物85g。减压蒸馏纯化得到(6-a)为无色油状液体60g,gc纯度为95%;从实施例1开始计算收率五步合计收率68%。
[0223]
实施例4:化合物(10-a)的合成
[0224]
步骤1:化合物(7-a)的合成
[0225][0226]
取实施例1步骤4所得的51.6g的(6-a)(0.27mol,1.0eq)用200ml四氢呋喃溶解备用。干燥除氧反应瓶内加入碘化亚铜0.5g(0.003mol,0.01eq),氮气保护下取联苯基溴化镁134ml(2.0m,1.0eq)加入反应体系,內温降至-15~-20℃后,取(6-a)的四氢呋喃溶液滴入反应体系,体系逐渐变为绿色,滴加完毕保持温度-5~-10℃反应1h。gc监测反应完毕,取50ml水淬灭反应,减压回收四氢呋喃后,加入二氯甲烷300ml和400ml水萃取反应,分液,有机相干燥脱溶得白色絮状固体,正庚烷打浆得到74g纯品(7-a),收率79%。
[0227]
(7-a)的表征数据:1h nmr(600mhz,chloroform-d)δ7.56(dd,j=19.0,7.5hz,4h),7.43(t,j=7.4hz,2h),7.33(dd,j=15.2,7.4hz,3h),4.85(d,j=6.8hz,1h),4.17(s,1h),3.65(d,j=10.0hz,1h),3.54(d,j=11.1hz,1h),2.94(dt,j=21.5,8.7hz,2h),1.44(s,9h).
13
c nmr(151mhz,cdcl3)δ155.03,140.75,139.72,136.13,129.71,128.76,127.36,127.23,126.98,79.84,51.98,46.96,37.41,28.33.
[0228]
表五:实施例4步骤1不同工艺条件筛选
[0229][0230]
碘化亚铜价格高,研究发现,碘化亚铜使用量为0.01eq时仍可有效催化反应,同时反应初步放大后反应效果与小试几乎一致。
[0231]
步骤2:化合物(10-a)的合成
[0232][0233]
取70g(0.2mol,1eq)的(7-a)加入200ml的甲苯,加热升温至110℃回流反应0.5h后,lc-ms监测(7-a)消失生成(8-a)。
[0234]
(8-a)的表征数据:1h nmr(400mhz,dmso-d6)δ7.83(s,1h),7.69-7.64(m,2h),7.61(d,j=8.2hz,2h),7.46(t,j=7.6hz,2h),7.40-7.31(m,3h),4.30(t,j=8.2hz,1h),4.15-4.05(m,1h),4.03(dd,j=8.2,5.4hz,1h),2.84(qd,j=13.6,6.1hz,2h);
13
c nmr(101mhz,dmso)δ159.11,140.35,138.87,136.34,130.49,129.40,127.79,127.14,127.00,68.57,52.95,40.35.
[0235]
取氢氧化钠24g(0.6mol,3eq)加入反应,保温80℃反应3h后lc-ms监测反应8-a消失生成(9-a);
[0236]
体系降温至25~30℃加入100ml水后滴加二碳酸二叔丁酯44g(0.2mol,1eq),体系逐渐变浑浊析出白色固体,滴加完毕后保温反应2h后hplc显示(9-a)消失生成(10-a),体系补加甲苯200ml加热升温至60℃搅拌30min,保温静置分层,弃去水相后有机相缓慢降温至0℃析出白色固体,过滤干燥得53g白色固体(10-a),纯度99.5%,三步合计收率85.5%。
[0237]
实施例5化合物(8-a)的合成
[0238]
步骤1:化合物(5-2-a)合成
[0239][0240]
实施例1步骤2的(4-a)的盐酸盐水溶液(0.374mol,1.0eq)加入2l反应瓶内,降温內温低于10℃后滴入氢氧化钠水溶液(17g氢氧化钠加入100ml水溶解,1.1eq)调节ph值为8~9之间,体系逐渐变为白色浑浊状态,滴加完毕,体系降温至0~10℃。取37g(1.1eq)氯甲酸甲酯用100ml二氯甲烷稀释后滴入反应体系,体系产生气体并放热,內温升至10~15℃,滴加完毕后保温反应2h后lc-ms监测反应原料消失,反应结束。静置分层,水相加入137g二氯甲烷萃取,分出有机相139g,合并有机相,122g水洗有机相,分出132g水相。有机相干燥过滤后减压浓缩得到淡黄色油状液体静置后变为白色蜡状固体105g为粗品(5-2-a)。
[0241]
(5-2-a)的表征数据:1h nmr(600mhz,chloroform-d)δ5.79(s,1h),4.89(s,1h),3.81(d,j=11.6hz,1h),3.73(dd,j=11.9,6.0hz,1h),3.68(s,3h),3.58(d,j=14.4hz,1h),3.54-3.47(m,1h),3.14(s,3h);
13
c nmr(151mhz,cdcl3)δ157.12,79.40,52.15,43.36,42.30,38.04.
[0242]
步骤2:化合物(6-2-a)的合成
[0243][0244]
取步骤3的105g(5-2-a)粗品(0.32mol,1eq)加入400ml四氢呋喃室温搅拌溶清,降温0~10℃,取60%的氢化钠19.2g(0.48mol,1.5eq)加入反应,体系产生气体泡沫,恢复室温25~30℃反应1h后lc-ms监测反应,原料消失。减压回收四氢呋喃后加入二氯甲烷,水萃取反应,有机相干燥脱溶得到58g淡黄色油状液体。减压蒸馏纯化得43.5g无色油状液体为(6-2-a),gc纯度95%,从实施例1开始计算前五步合计收率62.5%。
[0245]
(6-2-a)的表征数据:1h nmr(600mhz,chloroform-d)δ3.74(s,3h),3.65(dd,j=11.6,6.0hz,1h),3.51(dd,j=11.6,5.7hz,1h),2.82(dt,j=5.4,2.5hz,1h),2.47(d,j=6.0hz,1h),2.20(s,1h).
13
c nmr(151mhz,cdcl3)δ162.60,53.39,44.35,37.17,30.93.
[0246]
步骤3:化合物(7-2-a)的合成
[0247][0248]
取步骤2所得的43g的(6-2-a)(0.287mol,1.0eq)用200ml四氢呋喃溶解备用。
[0249]
干燥除氧反应瓶内加入碘化亚铜0.5g(0.0028mol,0.01eq),氮气保护下取联苯基溴化镁158ml(2.0m,1.1eq)加入反应体系,內温降至-15~-20℃后,取(6-a)的四氢呋喃溶液滴入反应体系,滴加完毕保持温度-10~-5℃反应1h。gc监测反应完毕,取50ml水淬灭反应,减压回收四氢呋喃后,加入二氯甲烷300ml和400ml水萃取反应,分液,有机相干燥脱溶得白色絮状固体,正庚烷打浆得到70g纯品(7-2-a),收率80%。
[0250]
(7-2-a)的表征数据:1h nmr(400mhz,chloroform-d)δ7.60-7.51(m,4h),7.43(t,j=7.6hz,2h),7.37-7.27(m,3h),5.03(d,j=7.4hz,1h),4.22(s,1h),3.68(s,3h),3.64(d,j=3.5hz,1h),3.54(dd,j=11.2,3.2hz,1h),2.96(t,j=6.4hz,2h);
13
c nmr(101mhz,cdcl3)δ156.19,140.63,139.81,135.82,129.63,128.75,127.40,127.25,126.95,52.48,52.24,46.71,37.30.
[0251]
步骤4:化合物(8-a)的合成
[0252][0253]
取步骤3中的(7-2-a)70g加入500ml反应瓶内,加入甲苯200ml加热升温110℃搅拌至回流2~3h,lc-ms监测反应(7-2-a)消失,生成(8-a),减压回收甲苯后得到浅黄色固体59.5g可直接用于下步反应。
[0254]
实施例6:化合物(8-a)的合成
[0255]
步骤1:化合物(5-3-a)的合成
[0256][0257]
实施例1步骤2的(4-a)的盐酸盐水溶液(0.374mol,1.0eq)加入2l反应瓶内,降温內温低于10℃后滴入氢氧化钠水溶液(17g氢氧化钠加入100ml水溶解,1.1eq)调节ph值为8~9之间,体系逐渐变为白色浑浊状态,滴加完毕,体系降温至0~10℃。取43.9g(1.1eq)氯甲酸乙酯用100ml二氯甲烷稀释后滴入反应体系,体系产生气体并放热,內温升至10~15℃,滴加完毕后保温反应2h后lc-ms监测反应原料消失,反应结束。静置分层,水相加入二氯甲烷萃取,分出有机相,合并有机相。有机相干燥过滤后减压浓缩得到淡黄色油状液体静置后变为白色蜡状固体112g为粗品(5-3-a)。
[0258]
(5-3-a)的表征数据:1h nmr(600mhz,chloroform-d)δ5.44(s,1h),4.90(s,1h),4.13(d,j=6.7hz,2h),3.80(d,j=12.2hz,1h),3.72(dd,j=12.2,6.0hz,1h),3.60(d,j=14.5hz,1h),3.51(dt,j=13.7,6.0hz,1h),3.13(s,3h),1.25(t,j=6.4hz,3h);
13
c nmr(151mhz,cdcl3)δ156.76,79.63,61.21,43.47,42.50,38.28,14.39.
[0259]
步骤2:化合物(6-3-a)的合成
[0260][0261]
取步骤3的112g(5-3-a)粗品(0.32mol,1eq)加入400ml四氢呋喃室温搅拌溶清,降温0~10℃,取60%的氢化钠26.18g(0.65mol,2eq)加入反应,体系产生气体泡沫,恢复室温25~30℃反应1h后lc-ms监测反应,原料消失。减压回收四氢呋喃后加入二氯甲烷,水萃取反应,有机相干燥脱溶得到54g淡黄色油状液体。减压蒸馏纯化得47.6g无色油状液体为(6-3-a),gc纯度94%,从实施例1开始计算前五步合计收率63%。
[0262]
步骤3:化合物(7-3-a)的合成
[0263][0264]
取步骤2所得的47g的(6-3-a)(0.287mol,1.0eq)用200ml四氢呋喃溶解备用。
[0265]
干燥除氧反应瓶内加入碘化亚铜0.5g(0.0028mol,0.01eq),氮气保护下取联苯基溴化镁158ml(2.0m,1.1eq)加入反应体系,內温降至-15~-20℃后,取(6-3-a)的四氢呋喃溶液滴入反应体系,滴加完毕保持温度-5~-10℃反应1h。gc监测反应完毕,取50ml水淬灭反应,减压回收四氢呋喃后,加入二氯甲烷300ml和400ml水萃取反应,分液,有机相干燥脱溶得白色絮状固体,正庚烷打浆得到77g纯品(7-3-a),收率85%。
[0266]
(7-3-a)的表征数据:1h nmr(400mhz,chloroform-d)δ7.60-7.51(m,4h),7.42(t,j=7.6hz,2h),7.32(dd,j=14.0,7.6hz,3h),5.01(d,j=7.7hz,1h),4.21(s,1h),4.12(q,j=7.1hz,2h),3.65(dd,j=10.8,3.8hz,1h),3.53(dt,j=11.1,3.6hz,1h),2.95(tt,j=
13.4,6.4hz,2h),1.24(t,j=7.1hz,3h);
13
c nmr(101mhz,cdcl3)δ155.78,140.64,139.76,135.89,129.63,128.73,127.36,127.22,126.93,61.04,52.35,46.75,28.29,14.51.
[0267]
步骤4:化合物(8-a)的合成
[0268][0269]
取步骤3中的(7-3-a)70g加入500ml反应瓶内,加入甲苯200ml加热升温110℃搅拌至回流2~3h,lc-ms监测反应(7-3-a)消失,生成(8-a),减压回甲苯后得到浅黄色固体63g可直接用于下步反应。
[0270]
实施例7化合物(10-a)的合成
[0271]
此实施例为公斤级放大反应,工艺室温均为25~35℃之间。
[0272]
步骤1:化合物(2-a)的合成
[0273][0274]
取苯甲醛1.0kg(9.42mol)加入乙醇4l室温搅拌,取氨水(25%)1l在温度15~20℃下滴入反应体系,滴加完毕恢复室温搅拌20min后,取(s)-环氧氯丙烷1.05kg(11.3mol,1.2eq)在氮气保护下用2l乙醇稀释后缓慢滴入反应体系,滴加完毕体系室温25~30℃搅拌反应12h。gc监测反应,环氧氯丙烷消失,反应结束。减压蒸馏回收乙醇得到黄色油状产品加入1l乙醇再次蒸馏后得到浅黄色油状液体2.3kg为粗品(2-a)。
[0275]
步骤2:化合物(4-a)的合成
[0276][0277]
取上步粗品(2-a)(2.3kg,8.5mol)加入8l二氯甲烷中,室温搅拌有少量白色不溶物,过滤,滤液加入三乙胺1.29kg(12.75mol,1.5eq),降温至內温0~10℃,滴加甲磺酰氯1.27kg(11.05mol,1.3eq),体系內温升至12~20℃,有白色物质析出。滴加完毕恢复室温搅拌1h后正相hplc监测反应,原料消失。体系加入2l水充分搅拌30min,萃取,有机相(2l
×
2)水洗涤两次。
[0278]
取1.2kg浓盐酸用2.4l水稀释后加入上步处理的有机相中,室温充分搅拌3~4h,静置分层,有机相用500ml水洗涤后合并水相得到粗品(4-a)水溶液直接用于下步反应。
[0279]
步骤3:化合物(5-a)的合成
[0280]
[0281]
步骤2(4-a)的盐酸盐水溶液(7.63mol)加入反应釜内,降温內温低于10℃后滴入氢氧化钠水溶液(460g氢氧化钠加入2l水溶解),体系逐渐变为白色浑浊状态,滴加完毕,体系降温至0~10℃。取1.7kg(7.63mol,1eq)二碳酸二叔丁酯滴入反应体系,体系产生气体并放热,內温升至10~15℃,滴加完毕后保温反应2h后lc-ms监测反应原料消失,反应结束。静置分层,反应加入2.7kg二氯甲烷萃取,静置分出有机相,2kg水洗有机相后有机相干燥过滤后减压蒸馏回收溶剂后得到淡黄色油状液体静置后变为白色蜡状固体2.5kg为粗品(5-a)。
[0282]
步骤4:化合物(6-a)的合成
[0283][0284]
取2.5kg的(5-a)粗品(7.65mol)加入5l四氢呋喃室温搅拌溶清备用。
[0285]
反应釜加入2l四氢呋喃,降温0~10℃,取60%的氢化钠460g(11.47mol,1.5eq)加入反应釜,保温搅拌30min后滴加(5-a)的四氢呋喃溶液,2~3h滴加完毕。升温至50℃反应5h后lc-ms监测反应,原料消失。500ml水淬灭反应后减压回收四氢呋喃,加入二氯甲烷4l,水2.5l萃取反应,有机相干燥脱溶得到1.75kg淡黄色油状液体。减压蒸馏纯化得1.1kg无色油状液体为6-a,gc纯度96%,前五步合计收率60%。
[0286]
步骤5:化合物(7-a)的合成
[0287][0288]
取步骤4所得的1.1kg的纯品(6-a)用4.5l四氢呋喃溶解备用,干燥除氧反应釜内加入碘化亚铜11g,氮气保护下取2.0m的联苯基溴化镁3l(6mol)加入反应体系,內温降至-15~-20℃后,取(6-a)的四氢呋喃溶液滴入反应体系,体系逐渐变为灰绿色,2h滴加完毕,保持温度反应1~2h。gc监测反应完毕,取2l水淬灭反应,减压回收四氢呋喃后,加入二氯甲烷5l和6l水萃取反应,分液,有机相干燥脱溶得白色絮状固体2.1kg,正庚烷打浆得到1.6kg纯品(7-a),收率79%。
[0289]
步骤6:化合物(10-a)的合成
[0290][0291]
取1.6kg的(7-a)加入8l的甲苯,加热升温至100~110℃回流反应1~2h后,lc-ms监测反应(7-a)消失生成(8-a)。降温补加甲苯2l后取氢氧化钠740g加入反应,回流反应3h后lc-ms监测反应(8-a)消失生成(9-a)。体系降温至25~30℃滴加二碳酸二叔丁酯1.1kg,体系逐渐变浑浊析出白色固体,滴加完毕后保温反应2h后hplc显示(9-a)消失生成(10-a),体系补加水2l后加热升温至50~60℃搅拌30min,保温静置分层,分出有机相并缓慢降温至0℃析出白色固体,过滤干燥得1.25kg白色固体(10-a),纯度99.3%,三步合计收率87%。
[0292]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的
限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1