一种培南类药物中间体的非对映异构体的合成方法
1.本发明涉及一种培南类药物中间体的非对映异构体的合成方法。
背景技术:
2.(3r,4r)-3-[(r)-1-叔丁基二甲基硅氧乙基]-4-乙酰氧基-2-氮杂环丁酮,以下简称为4aa,是合成培南类药物的关键中间体,在医药合成领域有着重要的用途。4aa结构有三个手性中心并且含有一个内酰胺环,立体选择性地构建内酰胺环是合成的关键与难点。在培南类抗生素的制备中,关键母核4aa具有非常重要的作用,所有的碳青霉烯类药物都要通过4aa来进一步反应制备。现有技术已知的制备4aa的方法有很多,其中,以苏氨酸为原料的路线一直以原料廉价、易得备受重视。
[0003]
在4aa的合成过程中,往往会产生一些影响药物纯度的物质,称之为杂质,包括有机杂质,无机杂质,残留溶剂等。杂质分析是药学研究的重要内容,同时涉及到药品的安全有效性。因此,合成及得到潜在杂质,对建立检测方法,分析杂质含量,并确定合理的杂质限度起到关键作用。4aa含有三个手性中心,因而有一个对映异构体杂质6个两两对映的非对映异构体杂质。本发明公布了一种4aa非对映异构体(3s,4s)-3-[(r)-1-叔丁基二甲基硅氧乙基]-4-乙酰氧基-2-氮杂环丁酮的合成方法,该非对映体构体结构如下:
[0004][0005]
(3s,4s)-3-[(r)-1-叔丁基二甲基硅氧乙基]-4-乙酰氧基-2-氮杂环丁酮
技术实现要素:
[0006]
本发明的目的是提供一种4aa非对映异构体(3s,4s)-3-[(r)-1-叔丁基二甲基硅氧乙基]-4-乙酰氧基-2-氮杂环丁酮的合成方法。
[0007]
本发明的技术方案如下:
[0008]
一种培南类药物中间体的非对映异构体10的合成方法,包括以下步骤:
[0009]
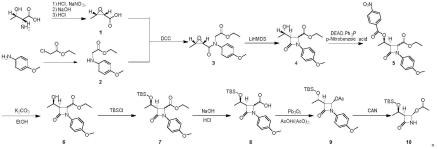
第一步:d-苏氨酸在盐酸与亚硝酸钠作用下发生重氮化、氯代,再于碱性物质作用下缩合(分子内亲核取代)得到化合物1;
[0010]
所述碱性物质为氢氧化钠;
[0011]
所述重氮化的温度为-5~5℃,并于该温度下一锅法完成氯代;所述碱性物质投料温度不超过5℃,并于室温(20~30℃)下完成缩合;
[0012]
第二步:对甲氧基苯胺与氯乙酸乙酯在碱性物质作用、100~120℃的条件下发生亲核取代反应得到化合物2;
[0013]
所述碱性物质为三乙胺;
2-氮杂环丁酮对建立4aa的检测方法,分析4aa的含量,并控制4aa生产中的杂质限度具有重要意义,为4aa的质量控制提供了重要保障。
附图说明
[0036]
图1(3s,4s)-3-[(r)-1-叔丁基二甲基硅氧乙基]-4-乙酰氧基-2-氮杂环丁酮1h nmr谱图。
具体实施方式
[0037]
下面通过具体实施例进一步描述本发明,但本发明的保护范围并不仅限于此。
[0038]
实施例1:中间体1化合物的制备
[0039]
在0℃下,将23.8g d-苏氨酸溶解于133ml 7.5m hcl,向体系中缓慢滴加40%(w/v)的22.8g亚硝酸钠水溶液并保持体系温度0℃(插温度计)。滴加完毕后,0℃下继续搅拌反应0.5小时后逐渐升温至室温,室温继续搅拌2小时。2小时后,加100ml水搅拌均匀,在0℃下缓慢滴加88ml 20%的氢氧化钠水溶液同时控制体系温度不超过5℃,滴毕,转移至室温反应3小时。反应完毕,0℃下,用7.5m hcl中和naoh并调节ph至1-2,用ea萃取后旋干得油状液体产物中间体1化合物17.5g,产率85.8%。
[0040]
实施例2:中间体2化合物的制备:
[0041]
在1l圆底烧瓶中,加入200ml三乙胺和24.6g对甲氧基苯胺,110℃下加热回流,缓慢滴加氯乙酸乙酯,滴加完毕后反应2小时。体系降温至80℃后可改搭蒸馏装置回收三乙胺,烧瓶壁上得到灰褐色固体,刮出后水洗,再用适量乙醇搅拌过夜,冷冻降温至0℃后灰黑色固体产物析出。过滤后滤饼用冷的乙醇:水=1:1洗涤并干燥得中间体2化合物34.7g,产率82.9%,避光保存。
[0042]
实施例3:中间体3化合物的制备
[0043]
将5.1g中间体1溶于75ml二氯甲烷中,在0℃下加入10.5g中间体2,搅拌十分钟,然后滴加15ml溶有10.3g n,n'-二环己基碳二亚胺的二氯甲烷溶液,继续搅拌2小时。反应完毕,旋干大部分溶剂后加入乙醚搅拌后冷冻至-10℃,再抽滤除去反应产生的二环己基脲。滤液旋干过柱,石油醚:乙酸乙酯=2:1洗脱,得到中间体3化合物11.8g,产率80.5%。
[0044]
实施例4:中间体4化合物的制备
[0045]
冰机冷却至-30℃,取三颈烧瓶充分干燥无水,将11.7g中间体3化合物加入,氮气保护后,加入150ml无水四氢呋喃,置于冰机中搅拌至反应体系温度降低到-30℃,缓慢滴加60ml双三甲基硅基胺基锂(1mol/l的四氢呋喃溶液),控制温度不高于-20℃,滴加完毕后冰机温度升至0℃,继续搅拌2小时。2m盐酸淬灭过量的双三甲基硅基胺基锂,乙酸乙酯萃取,柱层析纯化,石油醚:乙酸乙酯=1:1洗脱,得中间体4化合物9.8g,产率83.7%。
[0046]
实施例5:中间体5化合物的制备
[0047]
冰机降温到-15℃,将10g中间体4化合物、8.6g对硝基苯甲酸、15g三苯基膦加入到干燥的圆底烧瓶中,加入150ml无水thf,-15℃下滴加6.6ml偶氮二甲酸二乙酯。滴毕,缓慢升至室温,继续反应4小时。旋干溶剂,乙酸乙酯和水萃取,酯层旋干过柱,石油醚:乙酸乙酯=4:1洗脱。得中间体5化合物10.7g,产率71.2%。
[0048]
实施例6:中间体6化合物的制备
[0049]
将10g中间体5化合物溶于100ml无水乙醇中,加3.5g碳酸钾,室温剧烈搅拌8小时,旋干,乙酸乙酯和水萃取,合并有机相旋干,无需进一步处理直接用于下一步。
[0050]
实施例7:中间体7化合物的制备
[0051]
将所得的粗品中间体化合物6溶于10ml dmf中,0℃下加入5.1g叔丁基二甲基氯硅烷,室温搅拌过夜。乙酸乙酯和水萃取,有机相旋干后过柱纯化,石油醚:乙酸乙酯=1:1洗脱,得到中间体7化合物6.2g,两步产率67.4%。
[0052]
实施例8:中间体8化合物的制备
[0053]
将4.1g中间体7化合物溶于25ml甲醇中,滴加4.8ml 1m氢氧化钠水溶液,升温至50℃,反应3小时,旋干甲醇,用乙酸乙酯和水萃取,加入1m hcl调酸至ph=2,酯层旋干后得到中间体8化合物3.2g,产率84.4%。
[0054]
实施例9:中间体9化合物的制备
[0055]
在圆底烧瓶中加入10ml乙酸和10ml乙酸酐混合搅拌,加入8.3g红色四氧化三铅,升温至50℃下搅拌,待体系颜色由红色变为粉白色,加入5ml溶有3.8g中间体8化合物的乙酸溶液,保持温度继续反应2小时。旋干乙酸和乙酸酐,用乙酸乙酯和水萃取,有机相旋干,无需进一步纯化,得到中间体9化合物3.7g,产率96.7%。
[0056]
实施例10:式a化合物的制备
[0057]
取圆底烧瓶将3.9g中间体9化合物溶于100ml乙腈(或者溶剂也可以是四氢呋喃)中,将体系置于-20℃中,温度稳定后向体系中滴加硝酸铈铵的水溶液(50mmol,c=0.5mmol/ml),滴加完后搅拌30min。反应结束,加入亚硫酸钠搅拌淬灭醌,接着用碳酸氢钠洗涤,饱和食盐水洗涤,加入乙酸乙酯萃取,酯层合并后旋干得到棕色油状液体。过柱(pe:ea=1:1)纯化后得式10化合物2.3g,产率80.7%。
[0058]1h nmr(600mhz,cdcl3)δ6.50(s,1h),5.86(d,j=1.3hz,1h),4.27
–
4.22(m,1h),3.21(dd,j=3.4,1.3hz,1h),2.13(s,3h),1.27(d,j=6.3hz,3h),0.89(s,9h),0.09(d,j=8.1hz,6h)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1