一种双吖丙啶基光交联探针中间体及衍生物的制备方法
1.本发明属于有机合成技术领域,具体地说,涉及一种双吖丙啶基光交联探针中间体及衍生物的制备方法。
背景技术:
2.光交联标记技术结合了现代分子生物学、细胞生物学、有机化学等多学科的优势,运用合成的光敏小分子化合物作为工具探针,是分子水平上研究配体与受体相互作用的核心工具之一,对阐明配体与受体相互作用的机理以及药物先导物的发现有着巨大的推动作用。运用光交联标记技术,能够发现大量与疾病相关的靶点,确定多种小分子与生物大分子作用的模式,这些信息对合理设计药物具有非常重要的意义。
3.现有的靶标确证技术中通常需要在药物分子上连接较大体积的荧光基团来进行分析,这会导致探针分子出现活性降低、溶解度差、细胞渗透性差等缺点,光交联标记技术中常用的光交联基团,如苯基叠氮类、二苯甲酮类等基团的光交联活性并不高。随着光交联基团双吖丙啶的发现和生物正交反应技术的应用,现有技术中探针分子较大,光交联基团的光交联活性较低的问题得以解决。光交联基团双吖丙啶的体积很小,经特定波长的光照射后分解产生高活性的中间体,与结合受体的活性部位形成不可逆共价键,起到特异性标记的效果。
4.文献(z li,p hao,l li,e tal.angewandte chemie international edition,2013,125(33):8713-8718)公开结合了双吖丙啶光交联基团与生物正交反应基团的光交联探针,式(ii)所示化合物的合成与应用。式(ii)所示化合物通过式(i)所示化合物制备,在干冰浴的条件下,将式(i)所示化合物加入液氨,随后加入羟胺-o-磺酸的甲醇溶液,升至室温反应,过滤除去不溶物,滤液旋干后加入甲醇和三乙胺,在冰浴的条件下滴加碘的甲醇溶液,继续反应,萃取、洗涤、浓缩过柱得式(ii)所示化合物。式(ii)所示化合物可经碘代反应将羟基转化为碘,碘化物经两步反应可分别转化为羧基和氨基,通过这些基团与药物分子连接构成具有药物活性的光交联探针。
[0005][0006]
在光交联探针的合成路线中,式(i)所示双吖丙啶基光交联探针合成中间体的制
备尤为重要,是制备各种探针的必经之路。目前合成式(i)所示化合物的方法以上述文献所描述的方法为主,从乙酰乙酸乙酯出发,经过四步反应制备得到。过程中使用了正丁基锂、四氢铝锂等易燃易爆的危险试剂,容易引发事故,在安全、操作方面存在较大隐患;使用的底物炔丙基溴和溶剂苯都是易挥发且对人体具有较大危害的试剂,吸入或皮肤接触可能会引发中毒,增加工业化生产的操作难度;受底物乙酰乙酸乙酯的限制,该方法得到的产物碳数有限,难以根据需求获得不同长度的光交联探针;此外该方法需要四步完成,增加了操作工序,效率低。
技术实现要素:
[0007]
本发明的目的是提供一种安全、绿色、高效的双吖丙啶基光交联探针中间体及衍生物的制备方法。
[0008]
为了实现上述目的,本发明采用的技术方案如下:
[0009]
本发明的第一方面提供了一种双吖丙啶基光交联探针中间体及衍生物的制备方法,包括以下步骤:
[0010][0011]
将配体、过渡金属催化剂溶于溶剂中超声混匀,加入化合物iii、化合物ii、碱、光催化剂,在室温下充入氩气保护,光照反应,获得式i所示化合物;
[0012]
m选自0、1~10的整数(如1、2、3、4、5);n选自0、1~10的整数(如1、2、3、4、5);
[0013]
r选自羟基(oh)、-ch=ch2、氨基(-nh2)、酰胺(-conh2)、氯、氰基(-cn)、羧基(-cooh)、
[0014]
等。
[0015]
所述配体、过渡金属催化剂、化合物ii、碱、光催化剂与化合物iii的摩尔比为(0.01~0.2):(0.01~0.2):(0.1~5.0):(1~5.0):(0.001~0.1):1;优选为0.033:0.033:0.33:1:0.0067:1;
[0016]
所述化合物iii选自以下结构的一种:
[0017][0018]
所述化合物ii选自选自以下结构的一种:
[0019][0020]
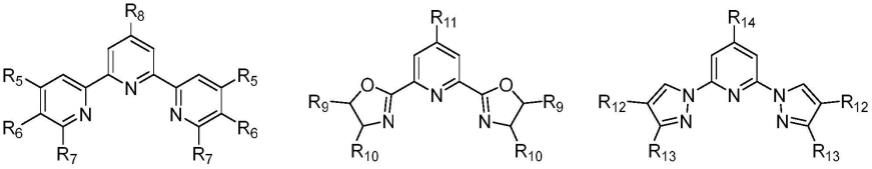
所述配体选自以下结构的一种:
[0021][0022]
r5、r6、r7、r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
各自独立的选自甲基、二甲基、乙基、二乙基、异丙基、叔丁基、1-金刚烷基、羧基、氰基、氨基、乙炔基、乙烯基、对甲苯基、对甲氧基苯基、4-吡啶基、对叔丁基苯基、苯基、苄基、cl、br、h;优选为4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶。
[0023]
所述过渡金属催化剂选自溴化镍、溴化镍六水合物、溴化镍乙烯二醇二甲基醚络合物、溴化镍二乙二醇二甲醚复合物、氯化镍、氯化镍乙二醇二甲基醚络合物、二乙酰丙酮镍、碘化镍;优选为溴化镍乙烯二醇二甲基醚络合物。
[0024]
所述溶剂选自丙酮、乙腈、二氯甲烷、水、二氯乙烷、硝基甲烷、二甲基亚砜,优选为丙酮。
[0025]
所述碱选自碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、磷酸钾、三乙胺、n,n-二异丙基乙胺;优选为碳酸钠。
[0026]
所述光催化剂选自四丁基铵十聚钨酸盐、十聚钨酸钠、十聚钨酸钾、蒽醌、ir[df(cf3)ppy]2(dtbbpy)pf6;优选为四丁基铵十聚钨酸盐。
[0027]
所述光照反应的波长范围为365~415纳米,优选为390纳米。
[0028]
所述光照反应的时间为1~24小时,优选为24小时;温度小于40℃,优选为35℃。
[0029]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0030]
本发明提供的双吖丙啶基光交联探针中间体及衍生物的制备方法,主要以醛与卤化物为主要原料,原料具有刺激性小、易于获得的特点。本发明在常温常压下,利用光催化偶联反应,合成步骤简洁,显著提高合成产率,且反应后除产物外生成无机盐,对环境污染小,绿色环保。本发明能够保证整个产品的质量得到进一步的提升,保证人们的生活得到进一步的改善,具有广阔的应用前景和市场需求。
[0031]
本发明的产率大幅提高,如实施例1与对比例1相比,收率提高了16%。与需要原位制备二异丙氨基锂的对比例1相比,实施例1的原料易于获得,且操作安全简便。对比例1中使用了-78℃或高温回流等温度变化较大的反应条件,在操作过程中和工业化上存在一定的困难,本发明实施例1中使用的条件为室温,条件温和,具有显著优势。对比例1的合成过程需要对酮羰基进行保护,使操作步骤的繁琐性增加,而实施例1兼容酮羰基,无需保护与脱保护步骤,使得效率大幅提高。对比例1中使用了大量毒性试剂如苯,大量危险试剂如正丁基锂,这些试剂都会为操作过程和工业化生产中带来极大的风险,实施例1中所使用的试剂绿色温和,对环境十分友好。
具体实施方式
[0032]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0033]
实施例1
[0034][0035]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(0.002mol,0.80g)、溴化镍乙烯二醇二甲基醚络合物(0.002mol,0.62g),加入100ml丙酮溶解,超声至溶液呈均一,依次向反应瓶中加入碳酸钠(0.06mol,6.36g)、四丁基铵十聚钨酸盐(0.0004mol,1.328g)、2-溴乙醇化合物ii-1(0.02mol,2.48g)、4-炔戊醛化合物iii-1(0.06mol,4.92g),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,24小时后加入100ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=2:1),得到2.09g式i-1所示化合物1-羟基-6-炔-3-庚酮。1h nmr(500mhz,cdcl3):δ3.87(t,j=5.0hz,2h),2.70(m,4h),2.47(t,j=7.2,2h),2.35(br,1h),1.96(t,j=2.7hz,1h);
13
c nmr(126mhz,cdcl3):δ208.57,82.58,68.92,57.65,43.98,41.16,12.57.hrms(m/z):[m+h]
+
calcd for c7h
11o2+
127.0681,found127.0682.
[0036]
实施例2
[0037][0038]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(0.002mol,0.80g)、溴化镍乙烯二醇二甲基醚络合物(0.002mol,0.62g),加入100ml丙酮溶解,超声至溶液呈均一,依次向反应瓶中加入碳酸钠(0.06mol,6.36g)、四丁基铵十聚钨酸盐(0.0004mol,1.328g)、3-溴丙醇化合物ii-2(0.02mol,2.76g)、6-炔庚醛化合物iii-2(0.06mol,5.76g),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,24小时后加入100ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=2:1),得到2.82g化合物i-2即1-羟基-8-炔-4-壬酮。1h nmr(500mhz,cdcl3):δ3.66(t,j=6.1hz,2h),2.62(d,j=6.9hz,2h),2.57(d,j=6.7hz,2h),2.24(qd,j=6.8,2.6hz,2h),1.97(t,j=2.7hz,1h),1.91(brs,1h),1.85(p,j=6.2hz,2h),1.80(p,j=7.0hz,2h);
13
c nmr(126mhz,cdcl3):δ210.9,83.5,69.1,62.2,41.1,39.6,26.4,22.2,17.7.hrms(m/z):[m+h]
+
calcd for c9h
15o2+
155.0994,found155.0992.
[0039]
实施例3
[0040][0041]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(0.002mol,0.80g)、溴化镍乙烯二醇二甲基醚络合物(0.002mol,0.62g),加入100ml丙酮溶解,超声至溶液呈均一,依次向反应瓶中加入碳酸钠(0.06mol,6.36g)、四丁基铵十聚钨酸盐(0.0004mol,1.328g)、4-溴-1-丁烯(0.02mol,2.70g)、4-炔戊
醛(0.06mol,4.92g),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,24小时后加入100ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=19:1),得到2.34g产物1-烯-8-炔-5-壬酮。1h nmr(500mhz,cdcl3):δ5.88-5.69(m,1h),5.08-4.89(m,2h),2.66(dd,j=8.0,6.5hz,2h),2.53(t,j=7.4hz,2h),2.45(ddd,j=9.4,6.5,2.7hz,2h),2.34(ddd,j=6.4,4.6,3.2hz,2h),1.94(t,j=2.7hz,1h);
13
c nmr(126mhz,cdcl3):δ207.7,136.9,115.4,83.1,68.7,41.8,41.3,27.6,12.9.hrms(m/z):[m+h]
+
calcd for c9h
13o+
137.0888,found 137.0889.
[0042]
实施例4
[0043][0044]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(0.002mol,0.80g)、溴化镍乙烯二醇二甲基醚络合物(0.002mol,0.62g),加入100ml丙酮溶解,超声至溶液呈均一,依次向反应瓶中加入碳酸钠(0.06mol,6.36g)、四丁基铵十聚钨酸盐(0.0004mol,1.328g)、(3-溴丙基)氨基甲酸叔丁酯(0.02mol,4.74g)、5-炔己醛(0.06mol,5.76g),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,24小时后加入100ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=8:1),得到3.69g产物(4-酮-8-炔-1-基)氨基甲酸叔丁酯。1h nmr(500mhz,cdcl3):δ4.59(br,1h),3.12(q,j=6.5hz,2h),2.57(t,j=7.2hz,2h),2.47(t,j=7.1hz,2h),2.23(td,j=6.8,2.7hz,2h),1.96(t,j=2.5hz,1h),1.79(p,j=7.1hz,2h),1.77(p,j=7.0hz,2h),1.44(s,9h);
13
c nmr(126mhz,cdcl3):δ209.8,156.0,83.5,79.2,69.1,41.1,39.9,28.4,24.1,22.2,17.7.hrms(m/z):[m+h]
+
calcd for c
14h25
no
3+
254.1756,found 254.1754.
[0045]
实施例5
[0046][0047]
在干燥反应瓶中加入磁力搅拌子,充入氩气保护,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(0.002mol,0.80g)、溴化镍乙烯二醇二甲基醚络合物(0.002mol,0.62g),加入100ml丙酮溶解,超声至溶液呈均一,依次向反应瓶中加入碳酸钠(0.06mol,6.36g)、四丁基铵十聚钨酸盐(0.0004mol,1.328g)、2-(2-溴乙基)-1,3-二氧戊环(0.02mol,3.58g)、5-炔己醛(0.06mol,5.76g),充入氩气保护。密封后在390纳米光源下光照,光源距离反应瓶约6厘米,风扇降温,控制温度为35℃,24小时后加入100ml丙酮稀释,减压浓缩,直接通过柱层析分离纯化(正己烷:乙酸乙酯=4:1),得到2.70g产物1-(1,3-二氧戊环-2-基)7-炔-3-辛酮。1h nmr(500mhz,cdcl3):δ4.88(t,j=4.3hz,1h),3.80-3.96(m,4h),2.55(t,j=5.9hz,2h),2.52(t,j=6.1hz,2h),2.20(td,j=6.9,2.6hz,2h),1.93-1.97(m,3h),1.73-1.80(m,2h);
13
c nmr(126mhz,cdcl3):δ209.3,103.2,83.6,68.9,64.9,41.0,36.5,27.5,22.2,17.7.hrms(m/z):[m+h]
+
calcd for c
11h17o3+
197.1099,found 197.1097.
[0048]
对比例1
[0049][0050]
向反应容器中加入二异丙基胺(50.6mmol,5.12g)和70mlthf,在氮气保护下降温至0℃或以下,滴加正丁基锂(50.6mmol,3.24g),搅拌后继续滴加乙酰乙酸乙酯(25.3mmol,3.29g),搅拌后再滴加炔丙基溴(25.3mmol,2.25ml),滴完后升至室温进行搅拌反应。随后倒入饱和氯化铵溶液中,萃取、洗涤、浓缩纯化,将所得产物(17.6mmol,2.96g)、对甲苯磺酸(1.34mmol,0.23g)、46ml苯和乙二醇(31.7mmol,1.8ml)混合后进行回流反应。分别用饱和碳酸氢钠、饱和氯化钠洗涤,浓缩纯化,所得产物(50.5mmol,11.0g)与300ml四氢呋喃混合后降温至0℃或以下,分批加入四氢铝锂(50.5mmol,1.92g),加完后升至室温进行搅拌反应。加入水淬灭,萃取、洗涤、浓缩纯化,所得产物(10mmol,1.7g)与30ml丙酮和对甲苯磺酸(2.5mmol,0.5g)混合后进行搅拌反应,浓缩纯化得到产物1-羟基-6-炔-3-庚酮。
[0051]
表1
[0052][0053][0054]
本发明的实施例1与对比例1相比,收率提高了16%。与需要原位制备二异丙氨基锂的对比例1相比,实施例1的原料易于获得,且操作安全简便。对比例1中需要使用-78℃或高温回流等温度变化较大的反应条件,会对操作过程和工业化带来困难,实施例1中使用的条件为室温,条件温和,具有显著优势。对比例1的合成过程需要对酮羰基进行保护,使操作步骤的繁琐性增加,而实施例1兼容酮羰基,无需保护与脱保护步骤,使得效率大幅提高。对比例1中使用了大量毒性试剂如苯,大量危险试剂如正丁基锂,这些试剂都会为操作过程和工业化生产中带来极大的风险,实施例1中所使用的试剂绿色温和,对环境十分友好。
[0055]
实施例6
[0056]
光催化剂的筛选:
[0057]
在4毫升干燥反应瓶中,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(10μmol,4.0毫克)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入0.5毫升丙酮,超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、光催化剂及所需的氢转移试剂(2.0μmol)、2-溴乙醇(0.1mmol,12.4毫克)、4-炔戊醛(0.3mmol,24.6毫克)。在室温
下,氩气保护,特定光源光照(十聚钨酸类光催化剂使用390纳米光源,ir[df(cf3)ppy]2(dtbbpy)pf6使用蓝色光源),风扇降温(控制温度低于40℃,约为35℃),反应24小时。
[0058]
表2
[0059]
光催化剂用量收率%四丁基铵十聚钨酸盐2.0μmol,6.6毫克,0.02当量83ir[df(cf3)ppy]2(dtbbpy)pf62.0μmol,2.0毫克,0.02当量59十聚钨酸钠2.0μmol,4.9毫克,0.02当量64四丁基铵十聚钨酸盐1.0μmol,3.3毫克,0.01当量63四丁基铵十聚钨酸盐5.0μmol,16.6毫克,0.05当量77
[0060]
使用四丁基铵十聚钨酸盐、ir[df(cf3)ppy]2(dtbbpy)pf6和十聚钨酸钠分别作为光催化剂,在相同用量(2.0μmol,0.02当量)下催化同一反应,使用四丁基铵十聚钨酸盐的收率最佳(83%)。降低和增加四丁基铵十聚钨酸盐的用量,都会导致收率降低(1.0μmol,0.01当量,63%;5.0μmol,0.05当量,77%)。最佳条件为四丁基铵十聚钨酸盐(2.0μmol,0.02当量)。
[0061]
实施例7
[0062]
过渡金属镍催化剂的筛选:
[0063]
在4毫升干燥反应瓶中,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(10μmol,4.0毫克)、过渡金属镍催化剂(10μmol),加入0.5毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、2-溴乙醇(0.1mmol,12.4毫克)、4-炔戊醛(0.3mmol,24.6毫克)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应24小时。
[0064]
表3
[0065]
过渡金属镍催化剂用量收率%溴化镍10μmol62溴化镍六水合物10μmol64镍溴化乙烯二醇二甲基醚络合物10μmol83溴化镍二乙二醇二甲醚复合物10μmol75氯化镍10μmol66氯化镍乙二醇二甲基醚络合物10μmol71二乙酰丙酮镍10μmol22碘化镍10μmol23
[0066]
分别使用各种过渡金属镍催化剂,在相同用量(10μmol)下催化同一反应,使用溴化镍乙烯二醇二甲基醚络合物的收率最佳(83%),其他溴化、氯化镍类催化剂也可以使用,碘化镍的收率不佳。最佳条件为溴化镍乙烯二醇二甲基醚络合物(10μmol)。
[0067]
实施例8
[0068]
配体的筛选:
[0069]
在4毫升干燥反应瓶中,依次加入配体(10μmol)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入0.5毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、2-溴乙醇(0.1mmol,
12.4毫克)、4-炔戊醛(0.3mmol,24.6毫克)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应24小时。
[0070]
表4
[0071]
配体用量收率%4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶10μmol834,4',4
”‑
三甲基-2,2':6',2
”‑
三联吡啶10μmol572,2':6',2
”‑
三联吡啶10μmol492,2'-联吡啶10μmol《5
[0072]
分别使用各种配体,在相同用量(10μmol)下催化同一反应,使用4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶的收率最佳(83%),其他配体也可以使用,收率相对较低,二齿类配体,如联吡啶不适合该反应。最佳条件为4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(10μmol)。
[0073]
实施例9
[0074]
碱的筛选:
[0075]
在4毫升干燥反应瓶中,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(10μmol,4.0毫克)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入0.5毫升丙酮(0.05m),超声溶解至溶液为均一,随后依次加入碱(0.30mmol)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、2-溴乙醇(0.1mmol,12.4毫克)、4-炔戊醛(0.3mmol,24.6毫克)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应24小时。
[0076]
表5
[0077][0078][0079]
分别使用各种碱,在相同用量(0.30mmol)下催化同一反应,使用碳酸钠的收率最佳(83%),碳酸氢钠、碳酸钾、碳酸氢钾也可以使用,磷酸钾、三乙胺、n,n-二异丙基乙胺的收率不佳,不适合使用。降低碳酸钠的用量,会导致收率降低(0.11mmol,48%;0.20mmol,58%)。最佳条件为碳酸钠(0.30mmol)。
[0080]
实施例10
[0081]
溶剂的筛选:
[0082]
在4毫升干燥反应瓶中,依次加入4,4',4
”‑
三叔丁基-2,2':6',2
”‑
三联吡啶(10μmol,4.0毫克)、溴化镍乙烯二醇二甲基醚络合物(10μmol,3.1毫克),加入溶剂,超声溶解至溶液为均一,随后依次加入碳酸钠(0.30mmol,31.8毫克)、四丁基铵十聚钨酸盐(2.0μmol,6.6毫克)、2-溴乙醇(0.1mmol,12.4毫克)、4-炔戊醛(0.3mmol,24.6毫克)。在室温下,氩气保护,390纳米光源光照,风扇降温(控制温度低于40℃,约为35℃),反应24小时。
[0083]
表6
[0084]
溶剂浓度收率%丙酮0.2m83乙腈0.2m《5二氯甲烷0.2m63水0.2m36二氯乙烷0.2m《5硝基甲烷0.2m《5二甲基亚砜0.2m23丙酮0.1m57
[0085]
分别使用各种溶剂,在相同浓度下(0.2m)下催化同一反应,使用丙酮的收率最佳(83%),二氯甲烷也可以使用,乙腈、水、二氯乙烷、硝基甲烷、二甲基亚砜的收率不佳,不适合使用。降低反应体系的浓度,会导致收率降低(0.1m,57%)。最佳条件为丙酮(0.2m)。
[0086]
以上所述仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专利的技术人员在不脱离本发明技术方案范围内,当可利用上述提示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1