一种磷酸可待因半水合物的制备方法与流程
1.本发明涉及药物合成技术领域,具体的,涉及一种磷酸可待因半水合物的制备方法。
背景技术:
2.磷酸可待因可形成三种水合物和两种无水物,而晶体结构的变化会改变固体的物理性质,而物理性质会影响更多特性,如:毒性、生物利用度、化学稳定性或保质期等。在磷酸可待因的所有晶型中,只有相差一个水分子的倍半水合物(c
18h21
no3·
h3po4·
1.5h2o)和半水合物(c
18h21
no3·
h3po4·
0.5h2o)在室温下是稳定的,因此只有倍半水合物和半水合物在商业上用于制备药物制剂。结晶水合物不同,存在一定质量差异,从目前数据考察上来看,在高温、光照情况下,倍半水合物失水较快,会很快转化为半水合物,最后变成无水i型,无水 ii型,这也是磷酸可待因倍半水合物长期放置易结块、发黄的主要原因。另外,从长期稳定性考察来看,不同结晶水合物有关物质增长不同,倍半水合物增长较快而半水合物有关物质加速实验过程中杂质无增长。综合以上两点,半水合物比倍半水合物更稳定。
3.目前,国外国内的可待因制剂目前均以倍半水合物为原料,可待因的合成方法同上述半水合物的合成路线一致即从cps粉中提取吗啡,再用三甲基苯基氯化铵作为甲化剂,碳酸钾作为碱,甲苯作为溶剂,将吗啡分子上的酚羟基甲基化获得可待因游离碱。其中结晶方法是经过在丙酮中成盐获得磷酸可待因倍半水合物,进而制备为所需制剂,而制剂的稳定性较差。因此,亟需开发一种磷酸可待因半水合物的合成工艺,以期提高磷酸可待因制剂产品的稳定性。
技术实现要素:
4.基于上述现有技术的缺陷,本发明提供一种磷酸可待因半水合物的制备方法,避免倍半水合物稳定性差的问题,进而提高制剂产品的质量。
5.为此,本发明提供一种磷酸可待因半水合物的制备方法,包括以下步骤:
6.可待因溶解于乙醇水溶液中,升温后滴加磷酸,待溶液ph为4.6-5.0时,降温养晶;养晶结束后滴加无水乙醇,继续降温,固液分离,干燥,得磷酸可待因半水合物。
7.优选的,可待因的质量与乙醇水溶液的体积比为2.5-4.0:1,所述质量与体积的比例关系为kg/l。
8.优选的,所述乙醇水溶液的体积百分数为78-82%。
9.优选的,可待因溶解后,升温至63-67℃。
10.优选的,所述磷酸与可待因的质量比为0.3-0.5:1。
11.优选的,滴加磷酸过程中,保持体系温度为66-70℃。
12.优选的,磷酸的体积百分数为40-45%。
13.优选的,降温养晶的温度为60-65℃,养晶时间为50-70min。
14.优选的,可待因的质量与无水乙醇的体积比为7-9:1,所述质量与体积的比值关系
为kg/l。
15.优选的,滴加无水乙醇的时间为110-130min。
16.优选的,滴加无水乙醇后,降温至≤10℃,降温维持时间≥120min。
17.本发明进一步提供一种上述制备方法得到的磷酸可待因半水合物。
18.本发明还提供一种用上述制备方法得到的磷酸可待因半水合物制备的可待因制剂,所述制剂为常规的药学制剂。
19.优选的,所述制剂选自片剂、口服液、注射液、糖浆中的一种或多种。
20.本发明的有益效果为:
21.1、本发明提供的磷酸可待因半水合物的制备方法,能够得到纯度大于99%的磷酸可待因半水合物。半水合物相比于倍半水合物更佳稳定,弥补了国内磷酸可待因半水合物制备工艺的空白,克服了现有的倍半水合物作原料稳定性较差的缺陷。
22.虽然倍半水合物在高温光照的条件下可转化为半水合物,但是直接通过高温处理,并不能保证得到的半水合物的纯度,其工艺条件难以控制,容易得到倍半水合物、半水合物、无水i型和无水ii型的混合物。而本发明通过选用乙醇作为溶剂,能够有效控制结晶,最终获得单一晶型的磷酸可待因半水合物,而非磷酸可待因的混晶。
23.2、本发明提供的磷酸可待因半水合物的制备方法具有工艺稳定,重现性好的特点。经过 3次小试、中式和规模化放大生产后制备得到磷酸可待因半水合物,分别进行质量检验,结果显示,其质量稳定。
附图说明
24.图1为实验例2中磷酸可待因半水合物有关物质检查的系统适应性色谱图;
25.图2为实验例2中lks170801批次产品的dsc图谱;
26.图3为实验例2中lks170802批次产品的dsc图谱;
27.图4为实验例2中lks170803批次产品的dsc图谱;
28.图5为实验例2中lks170801批次产品的tga图谱;
29.图6为实验例2中lks170802批次产品的tga图谱;
30.图7为实验例2中lks170803批次产品的tga图谱;
31.图8为实验例2中lks170801、lks170802和lks170803批次产品的xrpd图谱。
具体实施方式
32.下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都涉及本发明保护的范围。
33.实施例1
34.本实施例提供一种磷酸可待因半水合物的制备方法,包括以下步骤:
[0035] (1)向200l溶解罐中加入体积百分数为80%的乙醇溶液110l,打开搅拌,再加入30kg 可待因,盖好罐盖,搅拌20min使可待因溶解;
[0036] (2)将溶解后液体转入500l结晶罐中,升温至65℃后,开始滴加体积百分数为
43%的磷酸12kg,滴加过程中继续升温至68℃,测量反应液ph值,当ph=4.8时,降温至63℃;
[0037] (3)保持63℃养晶60min后,开始滴加无水乙醇240l,滴加时间为120min;
[0038] (4)滴加完毕后降温至8℃,在该温度下养晶3h,然后抽滤;滤饼投入到真空干燥机中,打开真空,通入60℃热水,进行干燥,得到磷酸可待因半水合物。
[0039]
实施例2
[0040]
本实施例提供一种磷酸可待因半水合物的制备方法,包括以下步骤:
[0041] (1)向200l溶解罐中加入体积百分数为78%的乙醇水溶液120l,打开搅拌,再加入 30kg可待因,盖好罐盖,搅拌30min使可待因溶解;
[0042] (2)将溶解后液体转入500l结晶罐中,升温至63℃后,开始滴加体积百分数为45%的磷酸9kg,滴加过程中继续升温至66℃,测量反应液ph值,当ph=4.6时,降温至60℃;
[0043] (3)保持60℃养晶70min后,开始滴加无水乙醇210l,滴加时间为110min;
[0044] (4)滴加完毕后降温至10℃,在该温度下养晶2h,然抽滤;滤饼投入到真空干燥机中,打开真空,通入60℃热水,进行干燥,得到磷酸可待因半水合物。
[0045]
实施例3
[0046]
本实施例提供一种磷酸可待因半水合物的制备方法,包括以下步骤:
[0047] (1)向200l溶解罐中加入体积百分数为82%的乙醇水溶液75l,打开搅拌,再加入30kg 可待因,盖好罐盖,搅拌10min使可待因溶解;
[0048] (2)将溶解后液体转入500l结晶罐中,升温至67℃后,开始滴加体积百分数为40%的磷酸15l,滴加过程中继续升温至70℃,测量反应液ph值,当ph=5.0时,降温至65℃;
[0049] (3)保持65℃养晶50min后,开始滴加无水乙醇270l,滴加时间为130min;
[0050] (4)滴加完毕后降温至5℃,在该温度下养晶2.5h,然后抽滤;滤饼投入到真空干燥机中,打开真空,通入60℃热水,进行干燥,得到磷酸可待因半水合物。
[0051]
实施例4
[0052]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用体积百分数为75%的乙醇溶解可待因。
[0053]
实施例5
[0054]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用体积百分数为85%的乙醇溶解可待因。
[0055]
实施例6
[0056]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)80%乙醇水溶液用量为150l。
[0057]
实施例7
[0058]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)80%乙醇水溶液用量为60l。
[0059]
实施例8
[0060]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(4)滴加完无水乙醇后降温至15℃。
[0061]
实施例9
[0062]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(2)滴加磷酸过程中,保持体系温度为60℃。
[0063]
实施例10
[0064]
本实施例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(2)滴加磷酸过程中,保持体系温度为75℃。
[0065]
对比例1
[0066]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用110l体积百分数为80%的丙酮溶解可待因。
[0067]
对比例2
[0068]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用110l体积百分数为80%的异丙醇溶解可待因。
[0069]
对比例3
[0070]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用110l体积百分数为80%的甲醇溶解可待因。
[0071]
对比例4
[0072]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用110l体积比为1:1的80%甲醇和80%乙醇混合溶液溶解可待因。
[0073]
对比例5
[0074]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(1)使用110l体积比为1:1:1的80%丙酮、80%甲醇和80%乙醇的混合溶液溶解可待因。
[0075]
对比例6
[0076]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(2)当ph=5.50时,开始降温。
[0077]
对比例7
[0078]
本对比例提供一种磷酸可待因半水合物的制备方法,其与实施例1相比,区别在于步骤(2)当ph=4.50时,开始降温。
[0079]
实验例1
[0080]
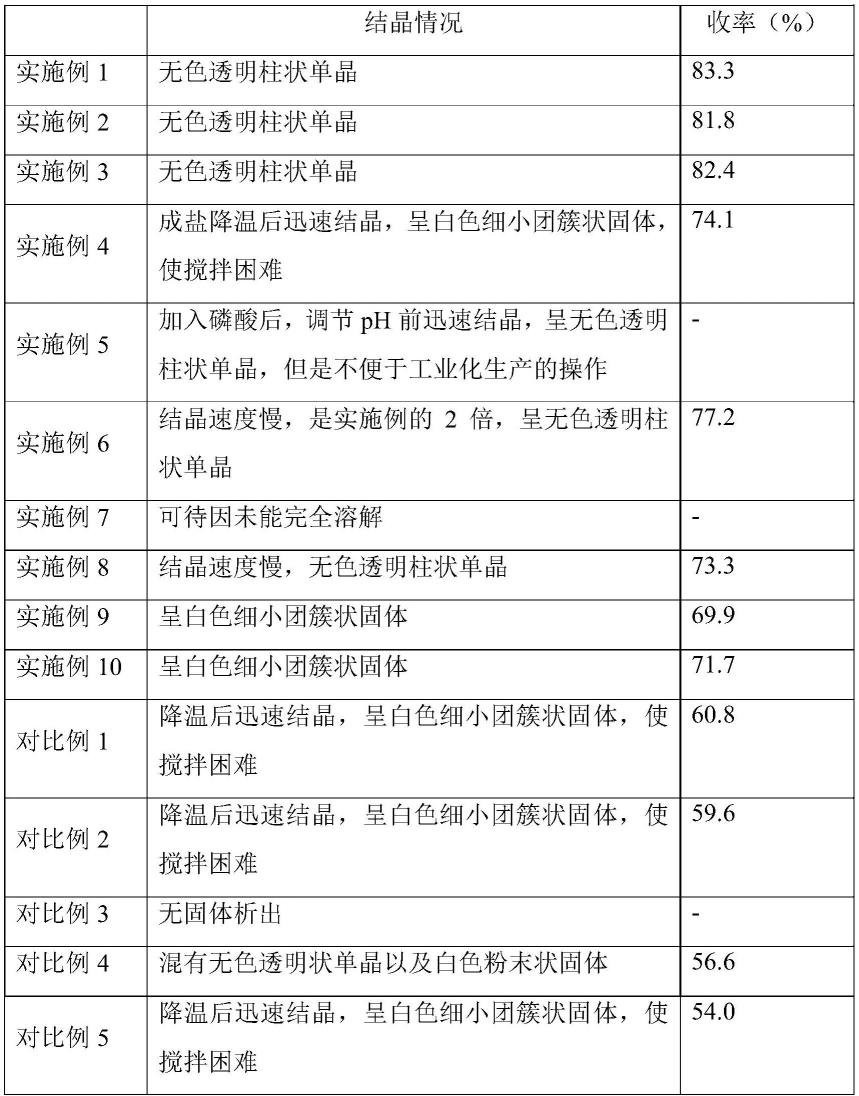
考察实施例1-10,对比例1-7的磷酸可待因半水合物结晶情况,以及最终产品的收率,结果见表1。
[0081]
表1不同工艺下磷酸可待因半水合物的结晶情况和收率
[0082][0083][0084]
由上表数据可以看出,本发明提供的磷酸可待因半水合物的生产方法,能够得到优质的晶体,并且收率较高。实施例4使用体积百分数为75%的乙醇,成盐降温后迅速结晶,呈白色细小团簇状固体,最终导致搅拌困难,对工业化生产上的控制产生一定不利影响;实施例 5使用体积百分数为85%的乙醇,可能乙醇量过多,导致其在加入磷酸溶液后便迅速
结晶,影响工艺控制。同理,实施例6和7在调整了乙醇的用量后,出现结晶速度慢以及溶解不完全等问题。实施例8降温养晶的温度较高,导致结晶情况较差,收率也较低。实施例9和10,控制可待因与磷酸成盐的温度过高或过低,最终晶型为白色细小团簇状固体,形状较差,而且收率也较低。
[0085]
对比例1-5分别采用不同种类的结晶溶剂,导致晶形、工业生产的控制以及收率都受到很大的影响。对比例6和对比例7未能严格控制加入磷酸后体系ph以及降温的时机,导致磷酸可待因晶型变化较大,收率较低。
[0086]
实验例2
[0087]
按照实施例1的方法重复3次,生产3个批次的磷酸可待因半水合物(lks170801-03),分别对生产出的半水合物进行有关物质和水分的分析,具体检测方法和结果如下:
[0088]
1.有关物质色谱条件:十八烷基硅烷键合硅胶为填充剂的色谱柱,以缓冲溶液(0.2%磷酸二氢钾1%庚烷磺酸钠的水溶液,磷酸调ph至3.2
±
0.2)-甲醇(7∶3)为流动相a;以流动相 a-乙腈(6∶4)作为流动相b,采用梯度洗脱:0~20min,100%a;40~45min,50%a;53min, 25%a;59min,0%a;59.5~66min,100%a。流速为1.0ml
·
min-1
,检测波长为245nm,柱温为40℃。
[0089]
表2有关物质检测结果
[0090][0091]
注:nd-未检出。
[0092]
杂质说明:
[0093]
①
原料引入:杂质b(吗啡)、g(蒂巴茵);
[0094]
②
工艺副产物:杂质a(甲基可待因);杂质c(可待因二聚体);杂质d(吗啡可待因二聚体);杂质f(14-羟基可待因)和杂质k(α-codeimethine);
[0095]
③
降解杂质:杂质e(10-羟基可待因)、杂质h(可待因氮氧化物)、杂质i(去甲可待因)、杂质j(可待因酮)。
[0096]
其中表格中未体现的杂质为未检出。
[0097]
2.水分
[0098]
2.1dsc检测条件:氮气气氛,升温速率10℃/分钟,升温范围30℃~300℃。结果见图2-4;
[0099]
2.2tga检测条件:氮气气氛,升温速率10℃/分钟,升温范围30℃~350℃。结果见图5-7;
[0100]
2.3xrpd检测条件:称取适量样品置于样品盘上,选用铜钯作为x-射线管靶,电压为 40千伏,电流为40毫伏,采用步径扫描的方式在3.000
°
到45.000
°
(2θ)的范围内进扫描。结果见图8。
[0101]
2.4水分检测:根据2020年版《中国药典》四部通则0832第一法(费休氏法)进行测
定,结果见表3。
[0102]
表3水分检测结果
[0103][0104]
表2数据表明,本发明提供的三批大生产磷酸可待因半水合物有关物质检出低,产品质量可靠;图2-8的dsc,tga,xrpd结果以及表3水分检测数据,表示三个批次产品的水分均为0.5结晶水,说明其制备工艺稳定,具有重复性和可靠性,适合大规模生产。
[0105]
实验例3
[0106]
考察本发明实施例1提供的磷酸可待因半水合物,市售磷酸可待因倍半水合物的稳定性,包括:
[0107]
①
外观:半水合物和倍半水合物置于相同环境中,倍水合物更易出现结块,发黄现象。
[0108]
②
有关物质:将半水合物和倍半水合物进行长期稳定性试验,二者0天和第36个月有关物质检测结果如下:
[0109]
表4半水合物和倍半水合物长期稳定性试验中有关物质含量变化
[0110][0111][0112]
上表数据表明,在长期稳定性试验中,倍半水合物中杂质增长略高于半水合物,说明半水合物稳定性更好。
[0113]
③
水分:
[0114]
表5半水合物和倍半水合物不同条件下水分检测结果
[0115][0116]
表5数据表面,产品包装完好,室温长期存放下水合物不会发生变化;在正常温度
日光照射下,1.5水合物会缓慢失水至接近0.5水状态。由此证明,半水合物稳定性优于倍半水合物。
[0117]
以上仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1